Expression Pattern of the Thioredoxin System in Human Endothelial Progenitor Cells and Endothelial Cells Under Hypoxic Injury
- Affiliations
-
- 1Division of Cardiology, Department of Internal Medicine, Chungbuk National University School of Medicine, Cheongju, Korea. mccho@chungbuk.ac.kr
- KMID: 2225164
- DOI: http://doi.org/10.4070/kcj.2010.40.12.651
Abstract
- BACKGROUND AND OBJECTIVES
The thioredoxin (TRx) system is a ubiquitous thiol oxidoreductase pathway that regulates cellular reduction/oxidation status. Although endothelial cell (EC) hypoxic damage is one of the important pathophysiologic mechanisms of ischemic heart disease, its relationship to the temporal expression pattern of the TRx system has not yet been elucidated well. The work presented here was performed to define the expression pattern of the TRx system and its correlation with cellular apoptosis in EC lines in hypoxic stress. These results should provide basic clues for applying aspects of the TRx system as a therapeutic molecule in cardiovascular diseases.
SUBJECTS AND METHODS
Hypoxia was induced with 1% O2, generated in a BBL GasPak Pouch (Becton Dickinson, Franklin Lakes, NJ, USA) in human endothelial progenitor cells (hEPC) and human umbilical vein endothelial cells (HUVEC). Apoptosis of these cells was confirmed by Annexin-V: Phycoerythrin flow cytometry. Expression patterns of TRx; TRx reductase; TRx interacting protein; and survival signals, such as Bcl-2 and Bax, in ECs under hypoxia were checked.
RESULTS
Apoptosis was evident after hypoxia in the two cell types. Higher TRx expression was observed at 12 hours after hypoxia in hEPCs and 12, 36, 72 hours of hypoxia in HUVECs. The expression patterns of the TRx system components showed correlation with EC apoptosis and cell survival markers.
CONCLUSION
Hypoxia induced significant apoptosis and its related active changes of the TRx system were evident in human EC lines. If the cellular impact of TRx expression pattern in various cardiovascular tissues under hypoxia or oxidative stress was studied meticulously, the TRx system could be applied as a new therapeutic target in cardiovascular diseases, such as ischemic heart disease or atherosclerosis.
Keyword
MeSH Terms
Figure
-
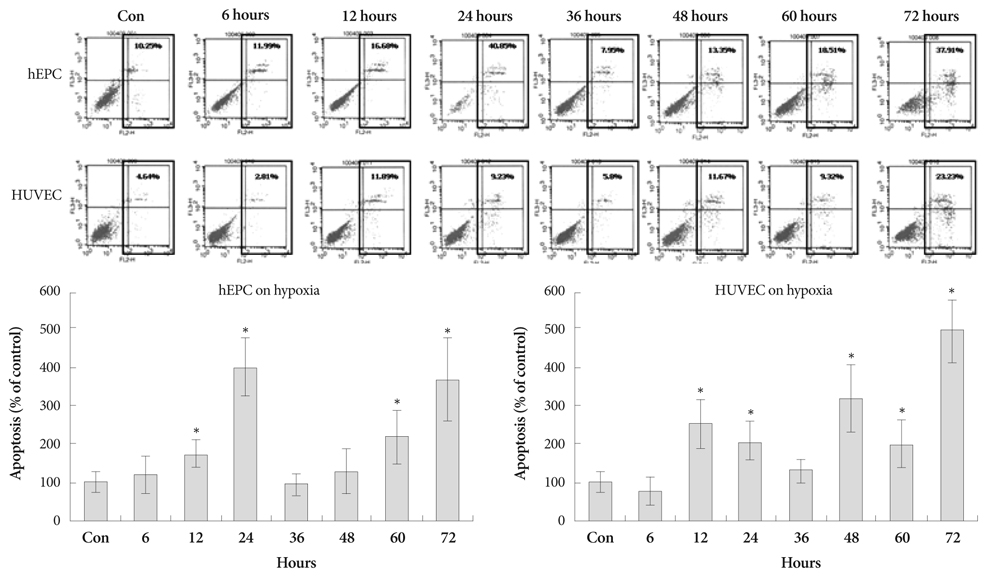
Fig. 1 Apoptosis in hEPC and HUVEC during hypoxia via FACS analysis. Hypoxia generated by the GasPak system, hEPC showed bimodal peaks of apoptosis at 12 and 24 hours (175.9±35.8%, 398.3±75.8%), 60 and 72 hours (220.5±68.1%, 368.8±20.3%) of hypoxia. HUVECs showed increased apoptosis at 12 hours (256.3±42.2%) of hypoxia initially and the highest degree of apoptosis was observed at 72 hours (500.6±45.6%) of hypoxia. *Significantly different from the control time (p<0.05). hEPC: human endothelial progenitor cells, HUVEC: human umbilical vein endothelial cells, FACS: fluorescence-activated cell sorting.
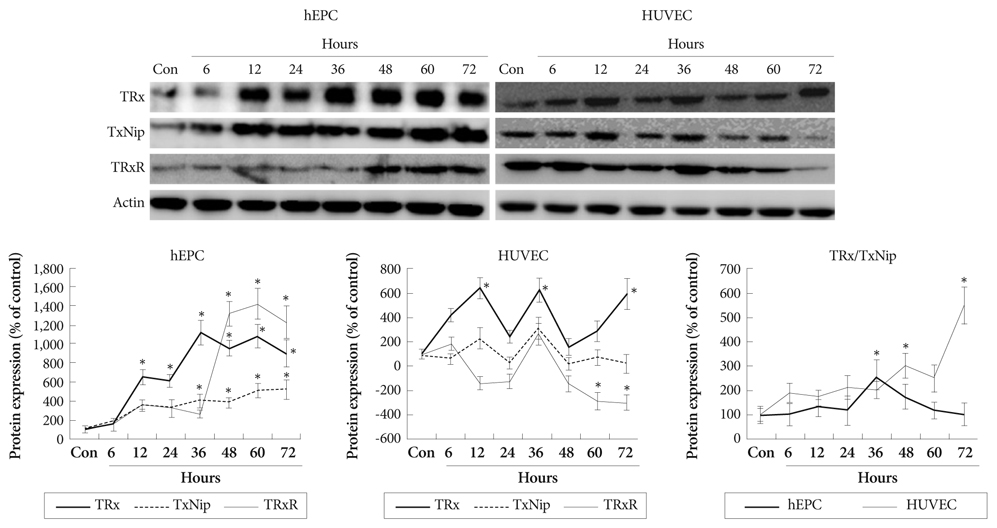
Fig. 2 The expression pattern of the TRx system in hEPCs and HUVECs during hypoxia. In hEPCs during hypoxia, TRx expression was elevated at 12 and 24 hours (651.3±78.5%, 593.5±64.3%) and thereafter. In HUVECs, TRx expression was increased at 12, 36, and 72 hours of hypoxia (640.1±89.4%, 620.2±98.1%, 595.9±128.4% on each time point), but the over-expression of TRxR and TxNip was not observed during whole hypoxic period. The TRx/TxNip ratio was increased at 36 hours (250.8±77.8%) of hypoxia in hEPCs and at 48 and 72 hours (301.7±49.2%, 550.4±78.4%) of hypoxia in HUVECs. *Significantly different from the control time (p<0.05). TRx: thioredoxin, hEPC: human endothelial progenitor cells, HUVEC: human umbilical vein endothelial cells, TRxR: TRx reductase, TxNip: TRx interacting protein.
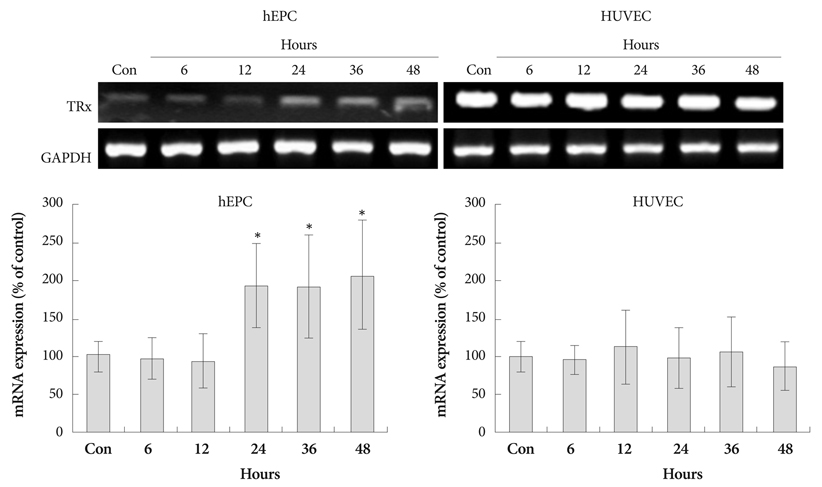
Fig. 3 TRx mRNA expression in hEPCs and HUVECs during hypoxic stress. TRx mRNA expression was increased significantly from 24 hours and thereafter during hypoxia in hEPCs (193.5±56.3%, 188.7±65.9%, 205.9±61.2% on 24, 36, 47 hours), but no increase in TRx mRNA expression in HUVECs was evident during the whole hypoxic period. *Significantly different from the control time (p<0.05). TRx: thioredoxin, mRNA: messenger ribonucleic acid, hEPC: human endothelial progenitor cells, HUVEC: human umbilical vein endothelial cells.
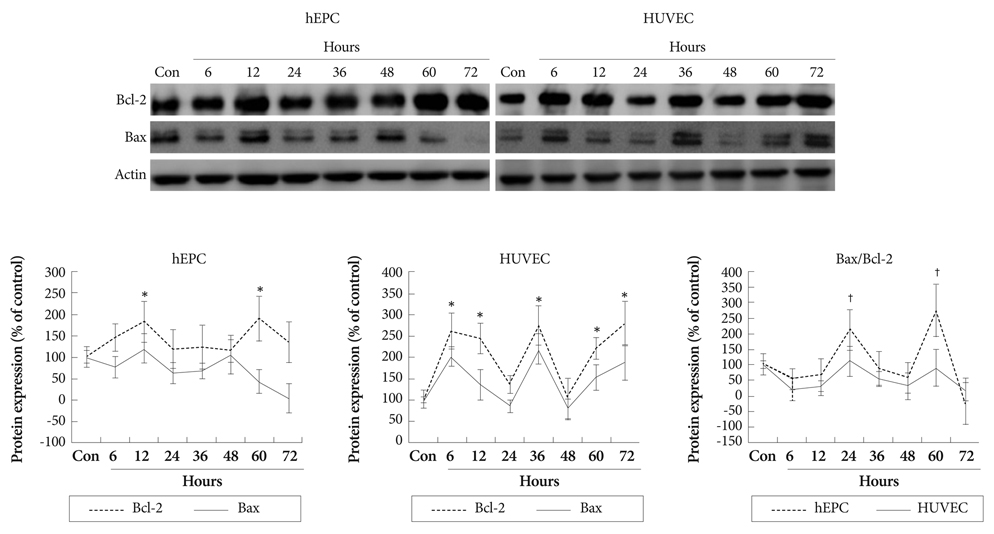
Fig. 4 The expression pattern of Bax and Bcl-2 in hEPCs and HUVECs during hypoxia. In hEPC, Bcl-2 expression showed 2 peaks at 12 and 60 hours (182.7±48.3%, 189.5±52.9%) of hypoxia, Bax expression was not increased significantly. In HUVECs, there were 3 peaks of Bcl-2 expression at 6 to 12, 36, and 60 to 72 hours (259.4±44.9%, 243.4±38.7%, 274.8±46.8%, 220.9±49.7%, 279.3±51.1% on each time point) of hypoxia and the Bax expression pattern was similar. Higher values of the Bax/Bcl-2 ratio were observed at 24 hours (212.4±67.0% vs. 110.8±48.3%) and 60 hours (275.9±86.3% vs. 89.5±59.1%) of hypoxia in hEPCs compared with HUVECs. *Significantly different from the control time (p<0.05), †Significantly different between hEPC and HUVEC (p<0.05). hEPC: human endothelial progenitor cells, HUVEC: human umbilical vein endothelial cells.
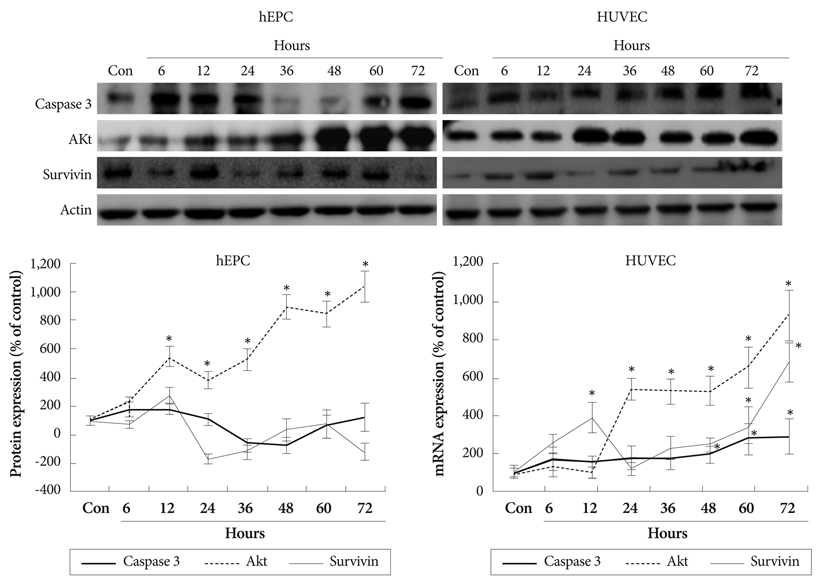
Fig. 5 The expression pattern of Caspase 3, Akt, and Survivin in hEPCs and HUVECs during hypoxia. A steep increase in the expression of Akt was observed in hEPC and HUVECs. Akt overexpression was significantly increased compared with control starting at 12 hours of hypoxia in hEPCs and starting at 24 hours of hypoxia in HUVECs. Caspase 3 and survivin activation was not evident in hEPCs during whole hypoxic period. Caspase 3 overexpression in HUVECs was observed starting at 48 hours (196.8±45.9%) of hypoxia. Higher survivin expression was observed at 12, 60, and 72 hours (385.7±75.2%, 345.6±89.1%, 678.7±95.7% on each time point) of hypoxia in hEPCs. *Significantly different from the control time (p<0.05). hEPC: human endothelial progenitor cells, HUVEC: human umbilical vein endothelial cells, mRNA: messenger ribonucleic acid.
Cited by 1 articles
-
Expression of Lectin like Oxidized Low Density Lipoprotein Receptor-1 in the Spontaneous Hypertensive Rat with High Cholesterol Diet
Ki-Seok Kim, No Kwan Park, Song-Yi Kim, Dong-Woon Kim, Seung-Jae Joo, Myeong-Chan Cho
J Korean Soc Hypertens. 2011;17(2):57-64. doi: 10.5646/jksh.2011.17.2.57.
Reference
-
1. Melley DD, Evans TW, Quinlan GJ. Redox regulation of neutrophil apoptosis and the systemic inflammatory response syndrome. Clin Sci. 2005. 108:413–424.2. Krohn K, Maier J, Paschke R. Mechanism of disease: hydrogen peroxide. DNA damage and mutagenesis in the development of thyroid tumors. Nat Clin Pract Endocrinol Metab. 2007. 3:713–720.3. Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: part I. basic mechanisms and in vivo monitoring of ROS. Circulation. 2003. 108:1912–1916.4. Sorescu D, Weiss D, Lassegue B, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002. 105:1429–1435.5. Brasen JH, Leppanen O, Inkala M, et al. Extracellular superoxide dismutase accelerates endothelial recovery and inhibits in-stent restenosis in stented atherosclerotic Watanabe heritable hyperlipidemic rabbit aorta. J Am Coll Cardiol. 2007. 50:2249–2253.6. Powis G, Briehl M, Oblong J. Redox signaling and the control of cell growth and death. Pharmacol Ther. 1995. 68:149–173.7. Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998. 273:6297–6302.8. Nonn L, Williams RR, Erickson RP, Powis G. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003. 23:916–922.9. Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997. 275:964–967.10. Masuda H, Kalka C, Asahara T. Endothelial progenitor cells for regeneration. Hum Cell. 2000. 13:153–160.11. Shioji K, Nakamura H, Masutani H, Yodoi J. Redox regulation by thioredoxin in cardiovascular diseases. Antioxid Redox Signal. 2003. 5:795–802.12. Haendeler J, Tischler V, Hoffmann J, Zeiher AM, Dimmeler S. Low doses of reactive oxygen species protect endothelial cells from apoptosis by increasing thioredoxin-1 expression. FEBS Lett. 2004. 577:427–433.13. Kalka C, Masuda H, Takahashi T, et al. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci U S A. 2000. 97:3422–3427.14. O'Donnell J, Mille-Baker B, Laffan M. Human umbilical-vein endothelial cells differ from other endothelial cells in failing to express ABO blood group antigens. J Vasc Res. 2000. 37:540–547.15. Gershoni JM, Palade GE. Electrophoretic transfer of proteins from sodium docedcyl sulfate-polyacrylamide gels to a positively charged mem-brane filter. Anal Biochem. 1982. 124:396–405.16. Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990. 323:27–36.17. Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events inpatients with coronary artery disease. Circulation. 2001. 104:2673–2678.18. Fridlyand LE, Philipson LH. Oxidative reactive species in cell injury: mechanisms in diabetes mellitus and therapeutic approaches. Ann N Y Acad Sci. 2005. 1066:136–151.19. Cho YS, Choi JH, Zhang SY, et al. Relationship of polymorphisms in the oxidative stress related genes-paraoxonase and p22phox to variant angina and coronary artery stenosis in Korean. Korean Circ J. 2003. 33:104–112.20. Kim KS, Han HS, Lee YS, et al. Plasma thioredoxin level and its correlation to myocardial damage in patients with acute myocardial infarction who underwent successful primary angioplasty. Korean Circ J. 2006. 36:39–45.21. Ide T, Tsutsui H, Kniganwa S, et al. Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res. 1999. 85:357–363.22. Powis G, Montfort WR. Properties and biological activities of thioredoxins. Annu Rev Pharmacol Toxicol. 2001. 41:261–295.23. Becker K, Gromer S, Schirmer RH, Muller S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur J Biochem. 2000. 267:6118–6125.24. Okuda M, Inoue N, Azumi H, et al. Expression of glutaredoxin in human coronary arteries: its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol. 2001. 21:1483–1487.25. Haendeler J, Hoffmann J, Zeiher AM, Dimmeler S. Antioxidant effects of statins via S-nitrosylation and activation of thioredoxin in endothelial cells: a novel vasculoprotective function of statins. Circulation. 2004. 110:856–861.26. Nishiyama A, Matsui M, Iwata S, et al. Identification of thioredoxin-binding protein-2/vitamin D(3) upregulated protein 1 as a negative regulator of thioredoxin function and expression. J Biol Chem. 1999. 274:21645–21650.27. Yoshioka J, Schulze PC, Cupesi M, et al. Thioredoxin-interacting protein controls cardiac hypertrophy through regulation of thioredoxin activity. Circulation. 2004. 109:2581–2586.28. World CJ, Yamawaki H, Berk BC. Thioredoxin in the cardiovascular system. J Mol Med. 2006. 84:997–1003.29. Bleeke T, Zhang H, Madamanchi N, Patterson C, Faber JE. Catecholoamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res. 2004. 94:37–45.30. Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008. 103:1072–1083.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- CD34 Expression in Pyogenic Granuloma
- Comparative Evaluation for Potential Differentiation of Endothelial Progenitor Cells and Mesenchymal Stem Cells into Endothelial-Like Cells
- Effects of Y-27632, a Rho-associated Kinase Inhibitor, on Human Corneal Endothelial Cells Cultured by Isolating Human Corneal Endothelial Progenitor Cells
- Endothelial progenitor cells and mesenchymal stem cells from human cord blood
- Endothelial Progenitor Cells' Classification and Application in Neurological Diseases
