Protective Effect of Survivin in Doxorubicin-Induced Cell Death in H9c2 Cardiac Myocytes
- Affiliations
-
- 1Graduate Program in Science for Aging, Yonsei University, Seoul, Korea.
- 2Cardiology Division, Severance Cardiovascular Hospital and Cardiovascular Research Institute, Seoul, Korea. smkang@yuhs.ac
- 3Brain Korea 21 Project for Medical Science, Yonsei University College of Medicine, Seoul, Korea.
- KMID: 2224901
- DOI: http://doi.org/10.4070/kcj.2013.43.6.400
Abstract
- BACKGROUND AND OBJECTIVES
Apoptosis has been known to be an important mechanism of doxorubicin-induced cardiotoxicity. Survivin, which belongs to the inhibitor of apoptosis protein family, is associated with apoptosis and alteration of the cardiac myocyte molecular pathways. Therefore, we investigated the anti-apoptotic effect and cellular mechanisms of survivin using a protein delivery system in a doxorubicin-induced cardiac myocyte injury model.
MATERIALS AND METHODS
We constructed a recombinant survivin which was fused to the protein transduction domain derived from HIV-TAT protein. In cultured H9c2 cardiac myocytes, TAT-survivin (1 microM) was added for 1 hour prior to doxorubicin (1 microM) treatment for 24 hours. Cell viability and apoptosis were evaluated by 2-(4,5-dimethyltriazol-2-yl)-2,5-diphenyl tetrazolium bromide assay, caspase-3 activity, and terminal deoxynucleotidyltransferase-mediated dUTP nick end-labeling assay. We measured the expression levels of several apoptosis-related signal proteins.
RESULTS
The survivin level was significantly reduced in a dose dependent manner up to 1 microM of doxorubicin in concentration. Purified recombinant TAT-survivin protein was efficiently delivered to H9c2 cardiac myocytes, and its transduction showed an anti-apoptotic effect, demonstrated by reduced caspase-3 activity and the apoptotic index, concomitantly with increased cell viability against doxorubicin injury. The phosphorylation of p38 mitogen-activated protein (MAP) kinase and the release of Smac from mitochondria were suppressed and the expression levels of Bcl-2 and cAMP response element-binding protein (CREB), the transcription factor of Bcl-2, were recovered following TAT-survivin transduction, indicating that survivin had an anti-apoptotic effect against doxorubicin injury.
CONCLUSION
Our results suggest that survivin has a potentially cytoprotective effect against doxorubicin-induced cardiac myocyte apoptosis through mechanisms that involve a decrease in the phosphorylation of p38 MAP kinase, mitochondrial Smac release, and increased expression of Bcl-2 and CREB.
Keyword
MeSH Terms
-
Apoptosis
Caspase 3
Cell Death
Cell Survival
Cyclic AMP Response Element-Binding Protein
Doxorubicin
Humans
Inhibitor of Apoptosis Proteins
Mitochondria
Myocytes, Cardiac
p38 Mitogen-Activated Protein Kinases
Phosphorylation
Phosphotransferases
Transcription Factors
Caspase 3
Cyclic AMP Response Element-Binding Protein
Doxorubicin
Inhibitor of Apoptosis Proteins
Phosphotransferases
Transcription Factors
p38 Mitogen-Activated Protein Kinases
Figure
-

Fig. 1 Effect of doxorubicin on cell viability and survivin level in H9c2 cardiac myocytes. A: H9c2 cardiac myocytes were treated with various concentrations of doxorubicin for 24 hours. Cell viability was assessed by MTT assay. The results present the means of three independent experiments. *p<0.05 compared to control. B: H9c2 cardiac myocytes were treated with doxorubicin for 24 hours. Equal amounts of protein were separated by SDS-PAGE gel, and immunoblot analysis was performed using anti-survivin antibody. C: cells were incubated with 1 µM of doxorubicin. At the indicated times, survivin levels were determined by immunoblot analysis. SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
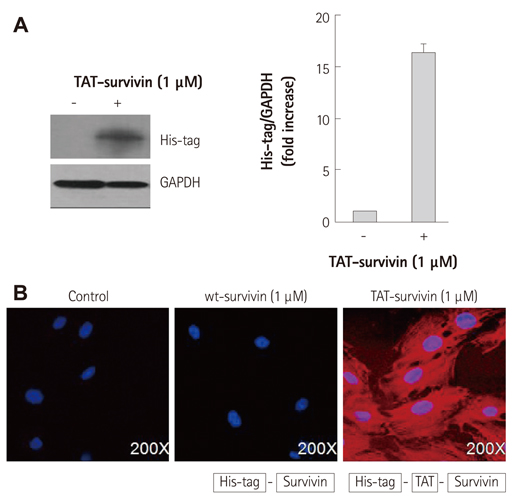
Fig. 2 Intracelluar delivery of recombinant TAT-survivin protein. A: H9c2 cardiac myocytes were treated with TAT-survivin protein for 1 hour. Immunoblot analysis was performed with anti-His-tag antibody. Normalized densitometric intensities of levels for transduced protein are shown as average fold changes. B: immunofluorescence microscopy analysis of H9c2 cardiac myocytes treated with 1 µM wt-survivin or TAT-survivin fusion proteins. After 1 hour, the cells were incubated with primary anti-His-tag antibody and Rhodamine-conjugated secondary antibody.
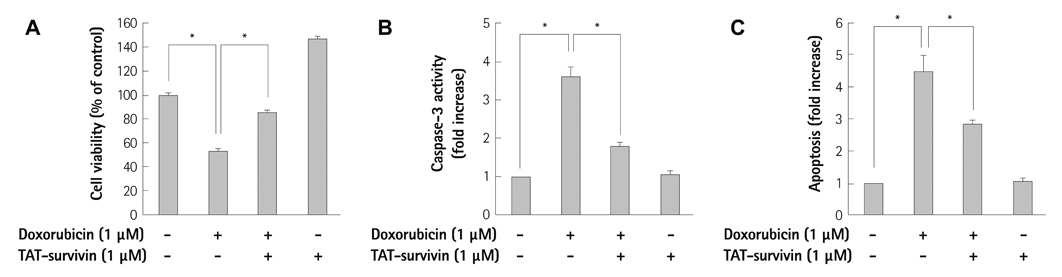
Fig. 3 Effect of TAT-survivin transduction on cell viability and apoptosis under doxorubicin treatment in H9c2 cardiac myocytes. The H9c2 cardiac myocytes were treated with 1 µM of TAT-survivin for 1 hour and then subjected to doxorubicin treatment for 24 hours. A: cell viability was assessed by the MTT assay. B: caspase-3 activity was determined using the caspase-3 activity assay kit. C: apoptotic cells were measured by the TUNEL assay. The results present the means of three independent experiments. *p<0.05.

Fig. 4 Effects of TAT-survivin on Bcl-2 expression in H9c2 cardiac myocytes. H9c2 cardiac myocytes were treated with 1 µM of TAT-survivin for 1 hour and then subjected to doxorubicin treatment for 24 hours. A: whole cell lysates were immunoblotted with anti-Bax or anti-Bcl-2 antibody. B: total RNA was purified from cells and subjected to RT-PCR using primers specific for Bcl-2. C: mitochondrial and cytoplasm fractions were prepared, and equal amounts of protein were separated by SDS-PAGE gel. The release of Smac and cytochrome C from mitochondria was detected using an anti-Smac and anti-cytochrome C antibody. D: equal amounts of nuclear protein were separated by SDS-PAGE gel, and immunoblot analysis was performed using anti-phospho-CREB or anti-CREB antibody. E: whole cell lysates were immunoblotted with anti-phospho-p38 or anti-p38 antibody. SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis, RNA: ribonucleic acid, CREB: cyclic adenosine monophosphate response elements-binding protein.

Fig. 5 Schematic representation regarding the protective effect of survivin in doxorubicin-induced cardiac cell death. This figure is derived from our experimental findings and other previously reported studies. Pro-apoptotic signaling cascades induced by doxorubicin are shown as solid lines. Dotted lines represent pathways that are pro-survival or carry an anti-apoptotic signal. Protective proteins including survivin are illustrated by the gray boxes. PTD: protein transduction domain.
Reference
-
1. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007; 49:330–352.2. Venkatakrishnan CD, Tewari AK, Moldovan L, et al. Heat shock protects cardiac cells from doxorubicin-induced toxicity by activating p38 MAPK and phosphorylation of small heat shock protein 27. Am J Physiol Heart Circ Physiol. 2006; 291:H2680–H2691.3. Venkatesan B, Prabhu SD, Venkatachalam K, et al. WNT1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal. 2010; 22:809–820.4. Johnson ME, Howerth EW. Survivin: a bifunctional inhibitor of apoptosis protein. Vet Pathol. 2004; 41:599–607.5. Song Z, Yao X, Wu M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J Biol Chem. 2003; 278:23130–23140.6. Levkau B. Survivin signalling in the heart. J Mol Cell Cardiol. 2011; 50:6–8.7. Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011; 194:355–365.8. Rosa J, Canovas P, Islam A, Altieri DC, Doxsey SJ. Survivin modulates microtubule dynamics and nucleation throughout the cell cycle. Mol Biol Cell. 2006; 17:1483–1493.9. Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: patent review. Expert Opin Ther Pat. 2010; 20:1723–1737.10. Levkau B, Schäfers M, Wohlschlaeger J, et al. Survivin determines cardiac function by controlling total cardiomyocyte number. Circulation. 2008; 117:1583–1593.11. Vivès E, Richard JP, Rispal C, Lebleu B. TAT peptide internalization: seeking the mechanism of entry. Curr Protein Pept Sci. 2003; 4:125–132.12. Kwon JH, Kim JB, Lee KH, et al. Protective effect of heat shock protein 27 using protein transduction domain-mediated delivery on ischemia/reperfusion heart injury. Biochem Biophys Res Commun. 2007; 363:399–404.13. Sakamoto KM, Frank DA. CREB in the pathophysiology of cancer: implications for targeting transcription factors for cancer therapy. Clin Cancer Res. 2009; 15:2583–2587.14. Meller R, Minami M, Cameron JA, et al. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J Cereb Blood Flow Metab. 2005; 25:234–246.15. Wong JC, Bathina M, Fiscus RR. Cyclic GMP/protein kinase G type-Iα (PKG-Iα) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Iα kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J Cell Biochem. 2012; 113:3587–3598.16. Leibowitz B, Yu J. Mitochondrial signaling in cell death via the Bcl-2 family. Cancer Biol Ther. 2010; 9:417–422.17. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron. 2002; 35:605–623.18. Yune TY, Park HG, Lee JY, Oh TH. Estrogen-induced Bcl-2 expression after spinal cord injury is mediated through phosphoinositide-3-kinase/Akt-dependent CREB activation. J Neurotrauma. 2008; 25:1121–1131.19. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta. 2010; 1802:396–405.20. Li P, Cavallero S, Gu Y, et al. IGF signaling directs ventricular cardiomyocyte proliferation during embryonic heart development. Development. 2011; 138:1795–1805.21. Ha JH, Noh HS, Shin IW, Hahm JR, Kim DR. Mitigation of H2O2-induced autophagic cell death by propofol in H9c2 cardiomyocytes. Cell Biol Toxicol. 2012; 28:19–29.22. Van Laethem A, Van Kelst S, Lippens S, et al. Activation of p38 MAPK is required for Bax translocation to mitochondria, cytochrome c release and apoptosis induced by UVB irradiation in human keratinocytes. FASEB J. 2004; 18:1946–1948.23. Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol Biol Cell. 2003; 14:2071–2087.24. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38 MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007; 11:175–189.25. Sen P, Chakraborty PK, Raha S. Activation of p38MAPK by repetitive low-grade oxidative stress leads to pro-survival effects. Biochim Biophys Acta. 2007; 1773:367–374.26. See F, Thomas W, Way K, et al. p38 mitogen-activated protein kinase inhibition improves cardiac function and attenuates left ventricular remodeling following myocardial infarction in the rat. J Am Coll Cardiol. 2004; 44:1679–1689.27. Gaitanaki C, Mastri M, Aggeli IK, Beis I. Differential roles of p38-MAPK and JNKs in mediating early protection or apoptosis in the hyperthermic perfused amphibian heart. J Exp Biol. 2008; 211(Pt 15):2524–2532.28. Watson JL, Greenshields A, Hill R, et al. Curcumin-induced apoptosis in ovarian carcinoma cells is p53-independent and involves p38 mitogen-activated protein kinase activation and downregulation of Bcl-2 and survivin expression and Akt signaling. Mol Carcinog. 2010; 49:13–24.29. Zhang J, Bui TN, Xiang J, Lin A. Cyclic AMP inhibits p38 activation via CREB-induced dynein light chain. Mol Cell Biol. 2006; 26:1223–1234.30. Grethe S, Ares MP, Andersson T, Pörn-Ares MI. p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-x(L). Exp Cell Res. 2004; 298:632–642.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Effects of Cardiotrophin-1 on Adriamycin-Induced Apoptosis in H9c2 Cardiomyoblasts
- The Effect of Adriamycin on Ionic Currents in Single Cardiac Myocytes of the Rabbit
- Adriamycin Induced Apoptosis of H9c2 Cardiomyocytes via a Caspase-Independent Pathway
- Protective Effect of Probucol against Adriamycin-Induced Apoptosis in Cultured Rat Cardiac Myocytes
- Zinc-Induced Cell Death in H9c2 Cardiomyoblast cells
