LPS-stimulated Macrophage Activation Affects Endothelial Dysfunction
- Affiliations
-
- 1Department of Pharmacology, Chungnam National University, College of Pharmacy, Daejeon, Korea. kheo@cnu.ac.kr
- KMID: 2409051
- DOI: http://doi.org/10.4167/jbv.2018.48.1.23
Abstract
- Intestinal microbiota is involved in the atherosclerotic process by development of an atheromatous core with foam cells in carotid arteries. It has reported that lipopolysaccharide (LPS) from Escherichia coli localizes in human atherosclerotic plaque and causes inflammation via interaction with toll like receptor 4. However, there is no evidence that whether LPS-activated macrophages regulate endothelial cell (EC) function. We evaluated whether LPS-activated macrophage acts as one of the stimulants activating EC and its underlying signaling pathways. Using Western blotting and quantitative reverse transcription-polymerase chain reaction (qRT-PCR), we confirmed that intraperitoneal injection with LPS increases iNOS protein and inflammatory cytokine, TNF-α and IL-6 mRNA expressions. To determine whether LPS-mediated macrophage inflammatory condition affects EC activation and inflammation, human umbilical vein endothelial cells (HUVECs) were incubated with isolated peritoneal macrophages from LPS-injected mice. Interestingly, p90RSK Serine 380 phosphorylation and protein expression were significantly increased by macrophage treatment in EC. Messenger RNA levels of vascular cell adhesion molecule 1 and p90RSK was increased, but endothelial nitric oxide synthase was decreased. In addition, NF-κB promoter activity, which plays an important role in the pathogenesis of inflammation, was strongly enhanced by the macrophage treatment in EC. We further evaluated the effects of LPS on EC function in the mouse aorta using en face staining. In agreement with in vitro result, p90RSK expression was strongly increased in the steady laminar flow region of the mouse aorta in mice injected with LPS. Together, our study demonstrates that p90RSK might be a one of the major therapeutic candidates for the prevention of vascular diseases mediated by LPS.
Keyword
MeSH Terms
-
Animals
Aorta
Atherosclerosis
Blotting, Western
Carotid Arteries
Endothelial Cells
Escherichia coli
Foam Cells
Gastrointestinal Microbiome
Human Umbilical Vein Endothelial Cells
Humans
In Vitro Techniques
Inflammation
Injections, Intraperitoneal
Interleukin-6
Macrophage Activation*
Macrophages*
Macrophages, Peritoneal
Mice
Nitric Oxide Synthase Type III
Phosphorylation
Plaque, Atherosclerotic
RNA, Messenger
Serine
Toll-Like Receptor 4
Vascular Cell Adhesion Molecule-1
Vascular Diseases
Interleukin-6
Nitric Oxide Synthase Type III
RNA, Messenger
Serine
Toll-Like Receptor 4
Vascular Cell Adhesion Molecule-1
Figure
-
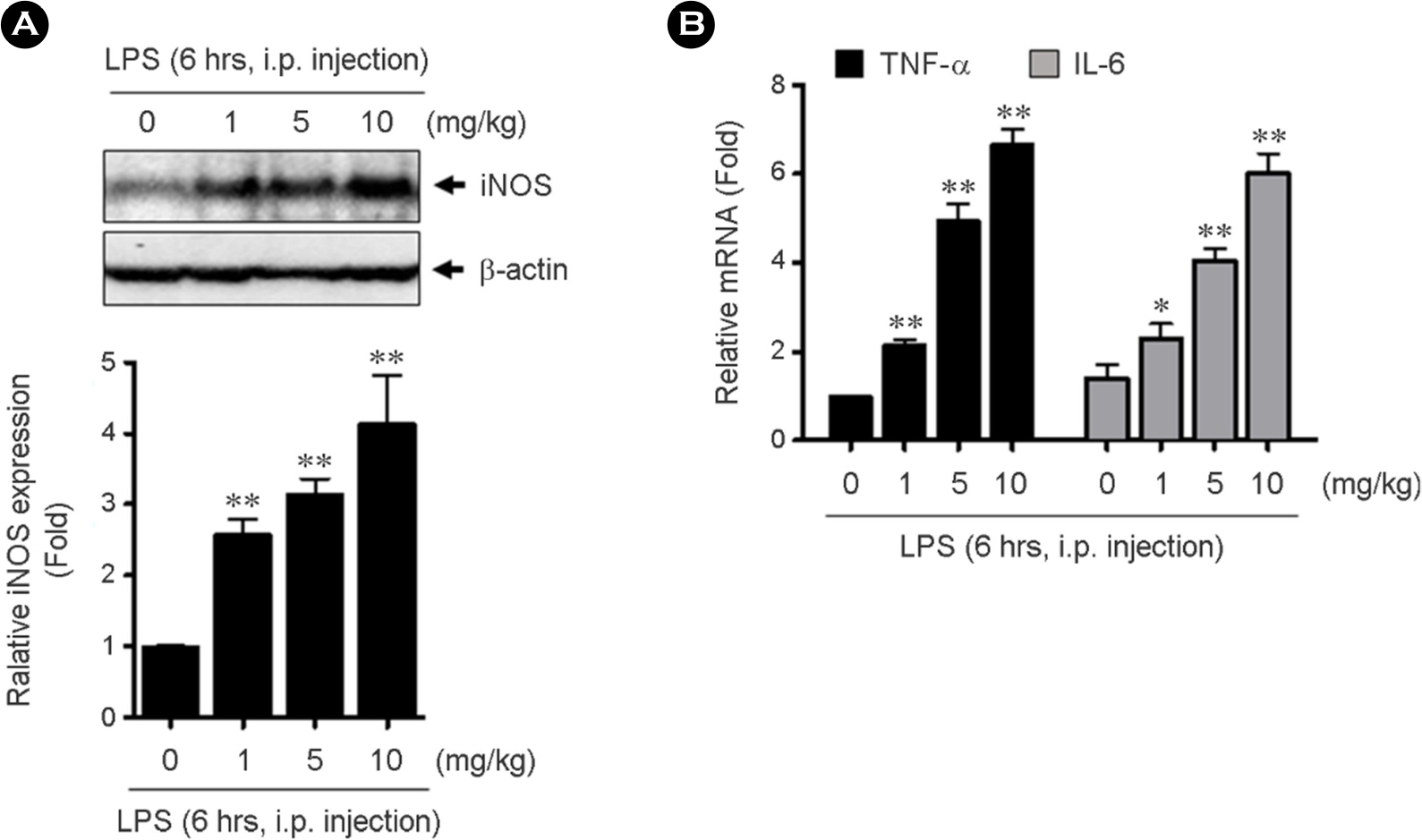
Figure 1. The expression of iNOS and inflammatory cytokines in LPS-activated peritoneal macrophages. LPS was i.p.injected with indicated doses in C57BL/6 mice. After 6 hrs, peritoneal macrophages were isolated and then assessed iNOS protein expression by western blotting using specific antibody (A) and TNF-α and IL-6 mRNAs by qRT-PCR using specific primers (B). Bar graph represents the quantification of relative fold changes of iNOS protein expression compared to the control in three independent experiments (0, A, lower panel). Data present mean ± SEM. ∗p< 0.05 and ∗∗p< 0.01, compared with control (0, PBS treated) by 1-way ANOVA followed by Bonferroni's post hoc test.
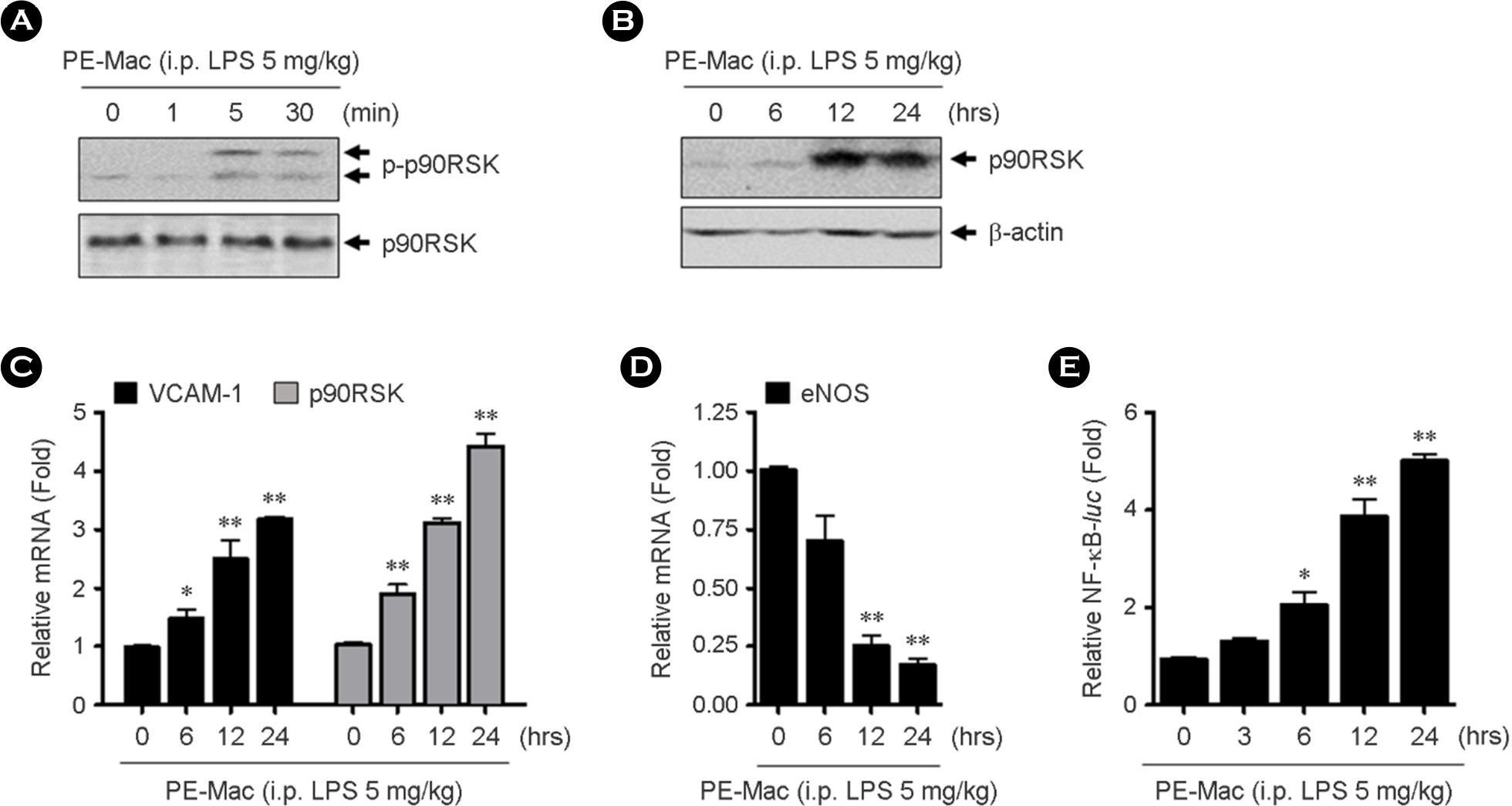
Figure 2. The effect of LPS-activated macrophages on EC functions. The HUVECs were co-cultured with macrophages isolated from 5 mg/kg LPS-injected mice for indicated times. (A-B) Phospho-p90RSK and total-p90RSK were determined by western blotting using specific antibody, respectively. (C-D) Total RNAs were isolated and qPT-PCR for VCAM-1, p90RSK, and eNOS mRNA levels were performed. (E) HUVECs were transfected with NF-κ B-lucand pRT-tkusing Lipofectamine 2000. The cells were co-cultured with the LPS-activated macrophages for indicated times and then promoter assay was performed. Data present mean ± SEM. ∗p< 0.05 and ∗∗p< 0.01, compared with control (0, PBS treated) by 1-way ANOVA followed by Bonferroni's post hoc test.
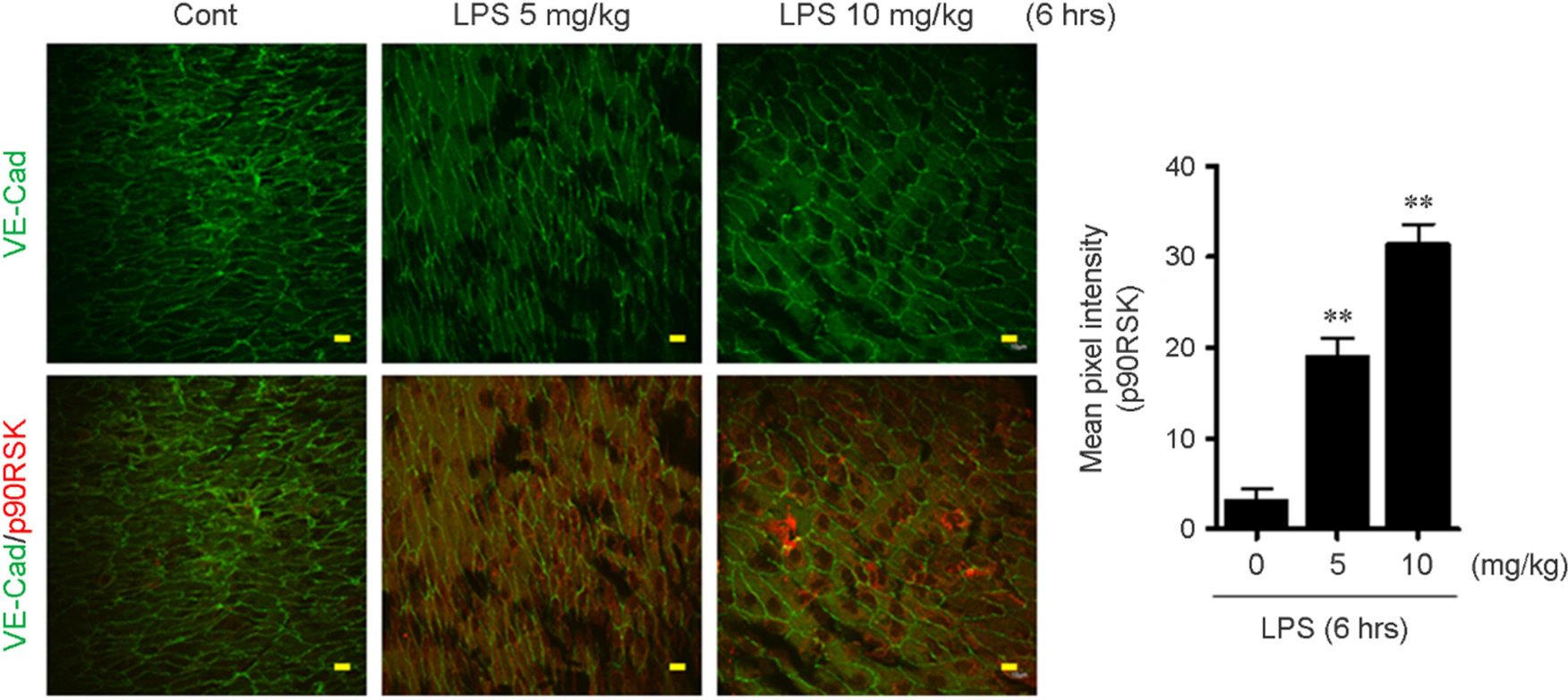
Figure 3. LPS stimulation increases p90RSK expression in aortic arch. After stimulation with 0 (PBS), 5, and 10 mg/kg LPS for 6 hrs, en facepreparations of the aortic arch of 7-week C57BL/6 mice were processed. The samples were double-stained with anti-VE-Cad (as an EC marker) and anti-p90RSK. Images were recorded using a confocal microscope equipped with a Planapo x60 1.42 NA oil objective lens. Scale bars: 10 μm. Bar graph indicates quantification of percentage of p90RSK fluorescence intensity. Data present mean ± SEM. ∗∗p< 0.01, compared with control (PBS treated) by 1-way ANOVA followed by Bonferroni's post hoc test.
Cited by 1 articles
-
Inhibitory Effect of Ginsenosides Rh1 and Rg2 on Oxidative Stress in LPS-Stimulated RAW 264.7 Cells
Yujin Jin, Naehwan Baek, Soyoung Back, Chang-Seon Myung, Kyung-Sun Heo
J Bacteriol Virol. 2018;48(4):156-165. doi: 10.4167/jbv.2018.48.4.156.
Reference
-
1). Li Q, Park K, Li C, Rask-Madsen C, Mima A, Qi W, et al. Induction of vascular insulin resistance and endothelin-1 expression and acceleration of atherosclerosis by the overexpression of protein kinase C-beta isoform in the endothelium. Circ Res. 2013; 113:418–27.2). Pucci ML, Miller KB, Dick LB, Guan H, Lin L, Nasjletti A. Vascular responsiveness to nitric oxide synthesis inhibition in hypertensive rats. Hypertension. 1994; 23:744–51.
Article3). Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, et al. Nitric oxide mediates angiogenesis in vivoand endothelial cell growth and migration in vitropromoted by substance P. J Clin Invest. 1994; 94:2036–44.4). Heo KS, Chang E, Le NT, Cushman H, Yeh ET, Fujiwara K, et al. De-SUMOylation enzyme of sentrin/SUMO-specific protease 2 regulates disturbed flow-induced SUMOylation of ERK5 and p53 that leads to endothelial dysfunction and atherosclerosis. Circ Res. 2013; 112:911–23.
Article5). Heo KS, Le NT, Cushman HJ, Giancursio CJ, Chang E, Woo CH, et al. Disturbed flow-activated p90RSK kinase accelerates atherosclerosis by inhibiting SENP2 function. J Clin Invest. 2015; 125:1299–310.
Article6). Blenis J. Signal transduction via the MAP kinases: proceed at your own RSK. Proc Natl Acad Sci U S A. 1993; 90:5889–92.
Article7). Ghoda L, Lin X, Greene WC. The 90-kDa ribosomal S6 kinase (pp90rsk) phosphorylates the N-terminal regulatory domain of IkappaBalpha and stimulates its degradation in vitro. J Biol Chem. 1997; 272:21281–8.8). Chen RH, Chung J, Blenis J. Regulation of pp90rsk phosphorylation and S6 phosphotransferase activity in Swiss 3T3 cells by growth factor-, phorbol ester-, and cyclic AMP-mediated signal transduction. Mol Cell Biol. 1991; 11:1861–7.
Article9). Fisher TL, Blenis J. Evidence for two catalytically active kinase domains in pp90rsk. Mol Cell Biol. 1996; 16:1212–9.
Article10). Xing J, Ginty DD, Greenberg ME. Coupling of the RAS-MAPK pathway to gene activation by RSK2, a growth factorregulated CREB kinase. Science. 1996; 273:959–63.
Article11). Le NT, Heo KS, Takei Y, Lee H, Woo CH, Chang E, et al. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013; 127:486–99.
Article12). Sanduzzi Zamparelli M, Compare D, Coccoli P, Rocco A, Nardone OM, Marrone G, et al. The Metabolic Role of Gut Microbiota in the Development of Nonalcoholic Fatty Liver Disease and Cardiovascular Disease. Int J Mol Sci. 2016; 17.
Article13). Lim JH, Jono H, Komatsu K, Woo CH, Lee J, Miyata M, et al. CYLD negatively regulates transforming growth factor-beta-signalling via deubiquitinating Akt. Nat Commun. 2012; 3:771.
Article14). Heo KS, Fujiwara K, Abe J. Disturbed-flow-mediated vascular reactive oxygen species induce endothelial dysfunction. Circ J. 2011; 75:2722–30.
Article15). Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, et al. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011; 193:867–84.16). McNeill E, Crabtree MJ, Sahgal N, Patel J, Chuaiphichai S, Iqbal AJ, et al. Regulation of iNOS function and cellular redox state by macrophage Gch1 reveals specific requirements for tetrahydrobiopterin in NRF2 activation. Free Radic Biol Med. 2015; 79:206–16.
Article17). Heiss C, Rodriguez-Mateos A, Kelm M. Central role of eNOS in the maintenance of endothelial homeostasis. Antioxid Redox Signal. 2015; 22:1230–42.
Article18). Kempe S, Kestler H, Lasar A, Wirth T. NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a proatherogenic program. Nucleic Acids Res. 2005; 33:5308–19.19). Sprague AH, Khalil RA. Inflammatory cytokines in vascular dysfunction and vascular disease. Biochem Pharmacol. 2009; 78:539–52.
Article20). Goveia J, Stapor P, Carmeliet P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med. 2014; 6:1105–20.
Article21). Jacobs AT, Ignarro LJ. Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. J Biol Chem. 2001; 276:47950–7.22). Cedergren J, Forslund T, Sundqvist T, Skogh T. Inducible nitric oxide synthase is expressed in synovial fluid granulocytes. Clin Exp Immunol. 2002; 130:150–5.
Article23). van Thienen JV, Fledderus JO, Dekker RJ, Rohlena J, van Ijzendoorn GA, Kootstra NA, et al. Shear stress sustains atheroprotective endothelial KLF2 expression more potently than statins through mRNA stabilization. Cardiovasc Res. 2006; 72:231–40.24). Heo KS, Berk BC, Abe J. Disturbed Flow-Induced Endothelial Proatherogenic Signaling Via Regulating Post-Translational Modifications and Epigenetic Events. Antioxid Redox Signal. 2016; 25:435–50.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Phosphatidylinositol-3-kinase Inhibitor Enhances Nitric Oxide Synthesis and Apoptosis in LPS-Stimulated Macrophages
- Silymarin Inhibits Morphological Changes in LPS-Stimulated Macrophages by Blocking NF-kappaB Pathway
- DICAM Inhibits Activation of Macrophage by Lipopolysaccharide
- Anti-inflammatory effects of rutin in lipopolysaccharide-stimulated canine macrophage cells
- The Regulatory Mechanism of Src Familly Kinase in Lipopolysaccharide (LPS) induced HF-kappaB Activation Pathway
