Glucose Oxidation and Production of Reactive Oxygen Species (ROS) in INS-1 Cells and Rat Islet Cells Exposed to High Glucose
- Affiliations
-
- 1Department of Internal Medicine, Yeungnam University College of Medicine, Korea.
- KMID: 2177594
- DOI: http://doi.org/10.4093/jkda.2006.30.4.246
Abstract
-
BACKGROUND: Chronic exposure of pancreatic islets to supraphysiologic concentrations of glucose causes beta cell dysfunction that is a process known as glucose toxicity. It has been reported that hyperglycemia increases the production of reactive oxygen species (ROS) in human islets and that ROS accumulation causes beta cell dysfunction associated with low capacity of intrinsic antioxidant enzymes. Also it has been postulated that this increase in ROS is prevented by an inhibitor of electron transport chain complex. The purpose of this study were to determine whether prolonged exposure of pancreatic islets to supraphysiologic glucose concentrations disrupts the intracellular balance between ROS thereby causing defective insulin secretion and to evaluate the site of hyperglycemia-induced ROS production.
METHODS
INS-1 cells & rat islets were incubated in increasing concentrations of glucose and either an inhibitor of complex I & II (TTFA), an uncoupler of oxidative phosphorylation (CCCP), aCCA, etc and also incubated in increasing concentration of glyceraldehyde and N-acetylcystein. Then intracellular peroxide levels by flow cytometric analysis and glucose induced insulin secretion were detected.
RESULTS
We observed that incubation with 30 mM glucose increased intracellular peroxide levels but decreased glucose-stimulated insulin secretion (GSIS) (P < 0.05). Exposure to TTFA, CCCP, aCCA did not reduce this increased intracellular peroxide levels, and did not increase GSIS (P < 0.05). 24-h incubation with glyceraldehyde at 5.6 mM glucose increased intracellular peroxide levels and decreased insulin content.
CONCLUSION
These observations indicate that there might be other origins in which ROS species are produced besides electron transport chain in mitochondria and glyceraldehyde may be a key molecule to produce ROS, and induce beta cell dysfunction.
Keyword
MeSH Terms
-
Animals
Carbonyl Cyanide m-Chlorophenyl Hydrazone
Diabetes Mellitus
Electron Transport
Glucose*
Glyceraldehyde
Humans
Hyperglycemia
Insulin
Islets of Langerhans*
Mitochondria
Oxidative Phosphorylation
Oxidative Stress
Rats*
Reactive Oxygen Species*
Carbonyl Cyanide m-Chlorophenyl Hydrazone
Glucose
Glyceraldehyde
Insulin
Reactive Oxygen Species
Figure
-

Fig. 1 Intracellular peroxide level (A, B) and GSIS (C, D) in INS-1 cells and rat islet cells after 3 days subculture. Data are mean ± SE from 3 separate experiments. *p < 0.05.

Fig. 2 Effects of several agents that affect mitochondrial metabolism on formation of hyperglycemia-induced intracellular peroxide in INS-1 cells. Cells were incubated in 5.6 mM glucose, 30 mM glucose alone, and 30 mM glucose plus either Mannoheptulose (MH), Methylsuccinate, aCCA (a-cyano-4-hydroxycinnamic acid), Rotenone, TTFA (Thenoyltrifluoroacetone), CCCP (Carbonyl cyanide chlorophenyl hydrazone), Azaserine, and intracellular peroxide levels were quantified. Data are mean ± SE from 3 separate experiments. *p < 0.05 vs control. **p < 0.05 vs 30 mM glucose.
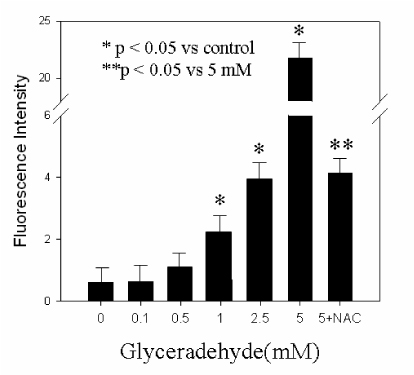
Fig. 3 Intracellular peroxide level in islets after 24 h culture with glyceraldehyde. Data are mean ± SE from 3 separate experiments. *p < 0.05 vs control. **p < 0.05 vs 5 mM.
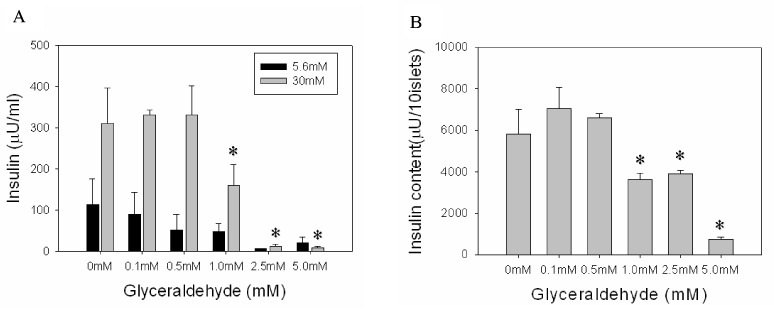
Fig. 4 GSIS (A) & Insulin content (B) in islets after 24 h culture with glyceraldehyde. Data are mean ± SE from 3 separate experiments. *p < 0.05 vs 0 mM.
Reference
-
1. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004. 279:42351–42354.2. Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990. 13:610–630.3. Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations J Cli. Invest. 1992. 90:320–325.4. Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest. 1993. 92:514–519.5. Robertson RP, Olson LK, Zhang HJ. Differentiating glucose toxicity from glucose desensitization: a new message from the insulin gene. Diabetes. 1994. 43:1085–1089.6. Poitout V, Olson LK, Robertson RP. Chronic Exposure of βTC-6 Cells to Supraphysiologic Concentrations of Glucose Decreases Binding of the RIPE3b1 Insulin Gene Transcription Activator. J Clin Inves. 1996. 97:1041–1046.7. Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999. 96:10857–10862.8. Baynes JW. Role of oxidative stress in development of complications in diabetes. Diabetes. 1991. 40:405–412.9. Kaneto H, Sharma A, Suzuma K, Laybutt DR, Bonner-Weir S, Weir GC. Induction of c-Myc Expression Suppresses Insulin Gene Transcription by Inhibiting NeuroD/ETA2-mediated Transcriptional Activation. J Biol Chem. 2002. 277:12998–13006.10. Kaneto H, Suzuma K, Sharma A, Bonner-Weir S, King GL, Weir GC. Involvement of Protein Kinase C β2 in c-myc Induction by High Glucose in Pancreatic β-Cells. J Biol Chem. 2002. 277:3680–3685.11. Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci U S A. 2000. 97:12222–12226.12. Wolff SP, Dean RT. Glucose autoxidation and protein modification. The potential role of 'autoxidative glycosylation' in diabetes. Biochem J. 1987. 245:243–250.13. Hunt JV, Dean RT, Wolff SP. Hydroxyl radical production and autoxidative glycosylation. Glucose autoxidation as the cause of protein damage in the experimental glycation model of diabetes mellitus and ageing. Biochem J. 1988. 256:205–212.14. Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000. 404:787–790.15. Kajimoto Y, Watada H, Matsuoka T, Kaneto H, Fujitani Y, Miyazaki J, Yamasaki Y. Suppression of transcription factor PDX-1/IPF1/STF-1/IDX-1 causes no decrease in insulin mRNA in MIN6 cells. J Clin Invest. 1997. 100:1840–1846.16. Olbrot M, Rud J, Moss LG, Sharma A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002. 99:6737–6742.17. Kataoka K, Han SI, Shioda S, Hirai M, Nishizawa M, Handa H. MafA is a glucose-regulated and pancreatic β-Cell-specific transcriptional activator for the insulin gene. J Biol Chem. 2002. 277:49903–49910.18. Matsuoka TA, Zhao L, Artner I, Jarrett HW, Friedman D, Means A, Stein R. Members of the large Maf transcription family regulate insulin gene transcription in islet Β Cells. Mol Cell Biol. 2003. 23:6049–6062.19. Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997. 46:1733–1742.20. Welsh N, Margulis B, Borg LA, Wiklund HJ, Saldeen J, Flodstrom M, Mello MA, Andersson A, Pipeleers DG, Hellerstrom C, et al. Differences in the expression of heat-shock proteins and antioxidant enzymes between human and rodent pancreatic islets: implications for the pathogenesis of insulin-dependent diabetes mellitus. Mol Med. 1995. 1:806–820.21. Hellman B, Idahl LA, Lernmark A, Sehlin J, Taljedal IB. The pancreatic beta-cell recognition of insulin secretagogues. Comparisons of glucose with glyceraldehyde isomers and dihydroxyacetone. Arch Biochem Biophys. 1974. 162:448–457.22. Taniguchi S, Okinaka M, Tanigawa K, Miwa I. Difference in mechanism between glyceraldehyde- and glucose-induced insulin secretion from isolated rat pancreatic islets. J Biochem(Tokyo). 2000. 127:289–295.23. MacDonald MJ, Mertz RJ, Rana RS. Glyceraldehyde phosphate: an insulin secretagogue with possible effects on inositol phosphate formation in pancreatic islets. Arch Biochem Biophys. 1989. 269:194–200.24. Alcazar O, Gine E, Qiu-Yue Z, Tamarit-Rodriguez J. The stimulation of insulin secretion by D-glyceraldehyde correlates with its rate of oxidation in islet cells. Biochem J. 1995. 310:215–220.25. Takahashi H, Tran PO, LeRoy E, Harmon JS, Tanaka Y, Robertson RP. D-Glyceraldehyde causes production of intracellular peroxide in pancreatic islets, oxidative stress, and defective beta cell function via non-mitochondrial pathways. J Biol Chem. 2004. 279:37316–37323.26. Lawrence MC, Bhatt HS, Watterson JM, Easom RA. Regulation of insulin gene transcription by a Ca(2+)-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol. 2001. 15:1758–1767.29. Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet. 1994. 344:721–724.30. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996. 20:463–466.32. Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A. 2002. 99:12363–12368.33. Du X, Matsumura T, Edelstein D, Rossetti L, Zsengeller Z, Szabo C, Brownlee M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J Clin Invest. 2003. 112:1049–1057.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Effect of Glucose on the Production of Reactive Oxygen Species in Retinal Pigment Epithelial Cells
- Effect of High Glucose on the Production of Reactive Oxygen Species in R28 Cells
- Effect of High Glucose on the Production of Reactive Oxygen Species in Trabecular Meshwork Cells
- Oxidative Stress of INS-1 Cell, HIT-T15 Cell and Rat Islet Cell as a Mechanism of Glucose Toxicity
- A Protective Role for Heme Oxygenase-1 in INS-1 Cells and Rat Islets that are Exposed to High Glucose Conditions
