Skeletal mineralization: mechanisms and diseases
- Affiliations
-
- 1Department of Bone and Mineral Research, Research Institute, Osaka Women's and Children's Hospital, Osaka Prefectural Hospital Organization, Izumi, Japan. michigami@wch.opho.jp
- KMID: 2468721
- DOI: http://doi.org/10.6065/apem.2019.24.4.213
Abstract
- Skeletal mineralization is initiated in matrix vesicles (MVs), the small extracellular vesicles derived from osteoblasts and chondrocytes. Calcium and inorganic phosphate (Pi) taken up by MVs form hydroxyapatite crystals, which propagate on collagen fibrils to mineralize the extracellular matrix. Insufficient calcium or phosphate impairs skeletal mineralization. Because active vitamin D is necessary for intestinal calcium absorption, vitamin D deficiency is a significant cause of rickets/osteomalacia. Chronic hypophosphatemia also results in rickets/osteomalacia. Excessive action of fibroblast growth factor 23 (FGF23), a key regulator of Pi metabolism, leads to renal Pi wasting and impairs vitamin D activation. X-linked hypophosphatemic rickets (XLH) is the most common form of hereditary FGF23-related hypophosphatemia, and enhanced FGF receptor (FGFR) signaling in osteocytes may be involved in the pathogenesis of this disease. Increased extracellular Pi triggers signal transduction via FGFR to regulate gene expression, implying a close relationship between Pi metabolism and FGFR. An anti-FGF23 antibody, burosumab, has recently been developed as a new treatment for XLH. In addition to various forms of rickets/osteomalacia, hypophosphatasia (HPP) is characterized by impaired skeletal mineralization. HPP is caused by inactivating mutations in tissue-nonspecific alkaline phosphatase, an enzyme rich in MVs. The recent development of enzyme replacement therapy using bone-targeting recombinant alkaline phosphatase has improved the prognosis, motor function, and quality of life in patients with HPP. This links impaired skeletal mineralization with various conditions, and unraveling its pathogenesis will lead to more precise diagnoses and effective treatments.
Keyword
MeSH Terms
-
Absorption
Alkaline Phosphatase
Calcium
Chondrocytes
Collagen
Diagnosis
Durapatite
Enzyme Replacement Therapy
Extracellular Matrix
Extracellular Vesicles
Familial Hypophosphatemic Rickets
Fibroblast Growth Factors
Gene Expression
Humans
Hypophosphatasia
Hypophosphatemia
Metabolism
Miners*
Osteoblasts
Osteocytes
Prognosis
Quality of Life
Receptors, Fibroblast Growth Factor
Rickets
Signal Transduction
Vitamin D
Vitamin D Deficiency
Alkaline Phosphatase
Calcium
Collagen
Durapatite
Fibroblast Growth Factors
Receptors, Fibroblast Growth Factor
Vitamin D
Figure
-
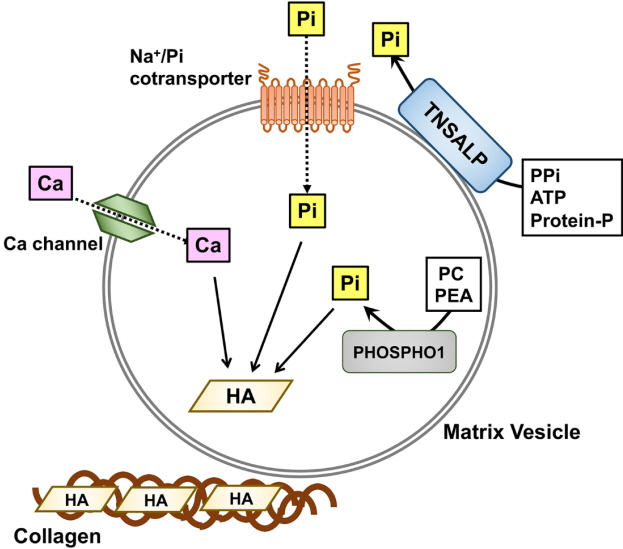
Fig. 1. MV-mediated skeletal mineralization. MVs are small vesicles derived from the plasma membrane of osteoblasts and chondrocytes. Tissue-nonspecific alkaline phosphatase (TNSALP) on the outer membrane of MVs hydrolyzes a mineralization inhibitor pyrophosphate [21], adenosine triphosphate (ATP), and protein-bound phosphate to produce Pi. Another phosphatase, PHOSPHO1 produces Pi from phosphocholine and phosphoethanolamine within MVs. Pi outside of MVs is transported into MVs partly by type III Na+ /Pi co-transporters PiT-1 and PiT-2. Calcium and Pi ions taken up by MVs crystallize to form hydroxyapatite, which subsequently propagates on collagen fibrils to mineralize the extracellular matrix. Pi, inorganic phosphate; PEA, phosphoethanolamine; PPi, pyrophosphate; PC, phosphocholine; HA, hydroxyapatite.
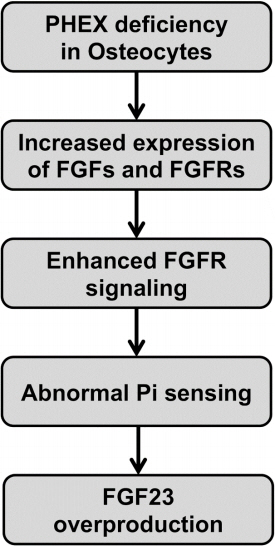
Fig. 2. Possible mechanism for FGF23 overproduction in the osteocytes of XLH. In PHEX-deficient osteocytes of XLH, enhanced FGFR signaling associated with increased FGFR1 expression may lead to abnormal Pi sensing and FGF23 overproduction. FGF23, fibroblast growth factor 23; XLH, X-linked hypophosphatemic ricket; FGRF, FGF receptor; PHEX, phosphate-regulating gene with homologies to endopeptidases, on the X chromosome; Pi, inorganic phosphate.
Reference
-
References
1. Young MF. Skeletal biology: where matrix meets mineral. Matrix Biol. 2016; 52-54:1–6.
Article2. Fukumoto S, Ozono K, Michigami T, Minagawa M, Okazaki R, Sugimoto T, et al. Pathogenesis and diagnostic criteria for rickets and osteomalacia--proposal by an expert panel supported by the Ministry of Health, Labour and Welfare, Japan, the Japanese Society for Bone and Mineral Research, and the Japan Endocrine Society. J Bone Miner Metab. 2015; 33:467–73.
Article3. Millán JL. What can we learn about the neural functions of TNAP from studies on other organs and tissues? Subcell Biochem. 2015; 76:155–66.
Article4. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12:233–46.
Article5. Millán JL. The role of phosphatases in the initiation of skeletal mineralization. Calcif Tissue Int. 2013; 93:299–306.
Article6. Michigami T, Ozono K. Roles of phosphate in skeleton. Front Endocrinol (Lausanne). 2019; 10:180.
Article7. Solomon DH, Browning JA, Wilkins RJ. Inorganic phosphate transport in matrix vesicles from bovine articular cartilage. Acta Physiol (Oxf). 2007; 190:119–25.
Article8. Yadav MC, Simão AM, Narisawa S, Huesa C, McKee MD, Farquharson C, et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. 2011; 26:286–97.
Article9. Yadav MC, Bottini M, Cory E, Bhattacharya K, Kuss P, Narisawa S, et al. Skeletal mineralization deficits and impaired biogenesis and function of chondrocyte-derived matrix vesicles in phospho1(-/-) and phospho1/Pi t1 double-knockout mice. J Bone Miner Res. 2016; 31:1275–86.
Article10. Goltzman D, Mannstadt M, Marcocci C. Physiology of the calcium-parathyroid hormone-vitamin D axis. Front Horm Res. 2018; 50:1–13.
Article11. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016; 96:365–408.
Article12. Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. 2016; 101:394–415.
Article13. Okazaki R, Ozono K, Fukumoto S, Inoue D, Yamauchi M, Minagawa M, et al. Assessment criteria for vitamin D deficiency/insufficiency in Japan - proposal by an expert panel supported by Research Program of Intractable Diseases, Ministry of Health, Labour and Welfare, Japan, The Japanese Society for Bone and Mineral Research and The Japan Endocrine Society [Opinion]. Endocr J. 2017; 64:1–6.
Article14. Michigami T, Kawai M, Yamazaki M, Ozono K. Phosphate as a signaling molecule and its sensing mechanism. Physiol Rev. 2018; 98:2317–48.
Article15. Mitchell HH, Hamilton TS, Steggerda FR, Bean HW. The chemical composition of the adult human body and its bearing on the biochemistry of growth. J Biol Chem. 1945; 158:625–37.
Article16. Murer H, Hildmann B. Transcellular transport of calcium and inorganic phosphate in the small intestinal epithelium. Am J Physiol. 1981; 240:G409–G416.
Article17. Sabbagh Y, O'Brien SP, Song W, Boulanger JH, Stockmann A, Arbeeny C, et al. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J Am Soc Nephrol. 2009; 20:2348–58.
Article18. Capuano P, Radanovic T, Wagner CA, Bacic D, Kato S, Uchiyama Y, et al. Intestinal and renal adaptation to a low-Pi diet of type II NaPi cotransporters in vitamin D receptorand 1alphaOHase-deficient mice. Am J Physiol Cell Physiol. 2005; 288:C429–C434.19. Beck L, Karaplis AC, Amizuka N, Hewson AS, Ozawa H, Tenenhouse HS. Targeted inactivation of Npt2 in mice leads to severe renal phosphate wasting, hypercalciuria, and skeletal abnormalities. Proc Natl Acad Sci U S A. 1998; 95:5372–7.20. Segawa H, Onitsuka A, Kuwahata M, Hanabusa E, Furutani J, Kaneko I, et al. Type IIc sodium-dependent phosphate transporter regulates calcium metabolism. J Am Soc Nephrol. 2009; 20:104–13.
Article21. Bergwitz C, Roslin NM, Tieder M, Loredo-Osti JC, Bastepe M, Abu-Zahra H, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am J Hum Genet. 2006; 78:179–92.
Article22. Bacic D, Lehir M, Biber J, Kaissling B, Murer H, Wagner CA. The renal Na+/phosphate cotransporter NaPi-IIa is internalized via the receptor-mediated endocytic route in response to parathyroid hormone. Kidney Int. 2006; 69:495–503.
Article23. Picard N, Capuano P, Stange G, Mihailova M, Kaissling B, Murer H, et al. Acute parathyroid hormone differentially regulates renal brush border membrane phosphate cotransporters. Pflugers Arch. 2010; 460:677–87.
Article24. Kovesdy CP, Quarles LD. Fibroblast growth factor-23: what we know, what we don't know, and what we need to know. Nephrol Dial Transplant. 2013; 28:2228–36.
Article25. Goetz R, Beenken A, Ibrahimi OA, Kalinina J, Olsen SK, Eliseenkova AV, et al. Molecular insights into the klothodependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol. 2007; 27:3417–28.
Article26. Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, et al. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006; 281:6120–3.
Article27. Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 2006; 444:770–4.
Article28. Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Kuro-o M, Mohammadi M, et al. The parathyroid is a target organ for FGF23 in rats. J Clin Invest. 2007; 117:4003–8.
Article29. Ohata Y, Yamazaki M, Kawai M, Tsugawa N, Tachikawa K, Koinuma T, et al. Elevated fibroblast growth factor 23 exerts its effects on placenta and regulates vitamin D metabolism in pregnancy of Hyp mice. J Bone Miner Res. 2014; 29:1627–38.30. Fukumoto S, Ozono K, Michigami T, Minagawa M, Okazaki R, Sugimoto T, et al. Pathogenesis and diagnostic criteria for rickets and osteomalacia - proposal by an expert panel supported by Ministry of Health, Labour and Welfare, Japan, The Japanese Society for Bone and Mineral Research and The Japan Endocrine Society. Endocr J. 2015; 62:665–71.
Article31. ADHR Consortium. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000; 26:345–8.32. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominant hypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab. 2011; 96:3541–9.
Article33. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A. 2011; 108:E1146–E1155.
Article34. A gene (PEX) with homologies to endopeptidases is mutated in patients with X-linked hypophosphatemic rickets. The HYP Consortium. Nat Genet. 1995; 11:130–6.35. Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan B, et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat Genet. 2006; 38:1310–5.
Article36. Lorenz-Depiereux B, Bastepe M, Benet-Pagès A, Amyere M, Wagenstaller J, Müller-Barth U, et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis. Nat Genet. 2006; 38:1248–50.
Article37. Lorenz-Depiereux B, Schnabel D, Tiosano D, Häusler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomalrecessive hypophosphatemic rickets. Am J Hum Genet. 2010; 86:267–72.
Article38. Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Höhne W, et al. Mutations in ENPP1 are associated with 'idiopathic' infantile arterial calcification. Nat Genet. 2003; 34:379–81.
Article39. Tagliabracci VS, Engel JL, Wen J, Wiley SE, Worby CA, Kinch LN, et al. Secreted kinase phosphorylates extracellular proteins that regulate biomineralization. Science. 2012; 336:1150–3.
Article40. Wang X, Wang S, Li C, Gao T, Liu Y, Rangiani A, et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012; 8:e1002708.
Article41. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A. 2014; 111:5520–5.
Article42. Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone. 2014; 67:56–62.
Article43. Rafaelsen SH, Raeder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20c mutations associated with fibroblast growth factor 23-related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. 2013; 28:1378–85.
Article44. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U S A. 2001; 98:6500–5.
Article45. Miyagawa K, Yamazaki M, Kawai M, Nishino J, Koshimizu T, Ohata Y, et al. Dysregulated gene expression in the primary osteoblasts and osteocytes isolated from hypophosphatemic Hyp mice. PLoS One. 2014; 9:e93840.
Article46. Bonewald LF. The amazing osteocyte. J Bone Miner Res. 2011; 26:229–38.
Article47. Martin A, Liu S, David V, Li H, Karydis A, Feng JQ, et al. Bone proteins PHEX and DMP1 regulate fibroblastic growth factor Fgf23 expression in osteocytes through a common pathway involving FGF receptor (FGFR) signaling. FASEB J. 2011; 25:2551–62.48. Xiao Z, Huang J, Cao L, Liang Y, Han X, Quarles LD. Osteocyte-specific deletion of Fgfr1 suppresses FGF23. PLoS One. 2014; 9:e104154.
Article49. White KE, Cabral JM, Davis SI, Fishburn T, Evans WE, Ichikawa S, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005; 76:361–7.
Article50. Yamazaki M, Ozono K, Okada T, Tachikawa K, Kondou H, Ohata Y, et al. Both FGF23 and extracellular phosphate activate Raf/MEK/ERK pathway via FGF receptors in HEK293 cells. J Cell Biochem. 2010; 111:1210–21.
Article51. Kimata M, Michigami T, Tachikawa K, Okada T, Koshimizu T, Yamazaki M, et al. Signaling of extracellular inorganic phosphate up-regulates cyclin D1 expression in proliferating chondrocytes via the Na+/Pi cotransporter Pit-1 and Raf/MEK/ERK pathway. Bone. 2010; 47:938–47.
Article52. Nishino J, Yamazaki M, Kawai M, Tachikawa K, Yamamoto K, Miyagawa K, et al. Extracellular phosphate induces the expression of dentin matrix protein 1 through the FGF receptor in osteoblasts. J Cell Biochem. 2017; 118:1151–63.
Article53. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician's guide to X-linked hypophosphatemia. J Bone Miner Res. 2011; 26:1381–8.
Article54. Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019; 7:189–99.
Article55. Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007; 2:40.
Article56. Mornet E, Nunes ME. Hypophosphatasia. In : Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, editors. GeneReviews((R)) [Internet]. Seattle (WA): University of Washington, Seattle;c2015. 1993–2019.57. Wenkert D, McAlister WH, Coburn SP, Zerega JA, Ryan LM, Ericson KL, et al. Hypophosphatasia: nonlethal disease despite skeletal presentation in utero (17 new cases and literature review). J Bone Miner Res. 2011; 26:2389–98.
Article58. Ozono K, Yamagata M, Michigami T, Nakajima S, Sakai N, Cai G, et al. Identification of novel missense mutations (Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. J Clin Endocrinol Metab. 1996; 81:4458–61.
Article59. Simon-Bouy B, Taillandier A, Fauvert D, Brun-Heath I, Serre JL, Armengod CG, et al. Hypophosphatasia: molecular testing of 19 prenatal cases and discussion about genetic counseling. Prenat Diagn. 2008; 28:993–8.
Article60. Michigami T, Uchihashi T, Suzuki A, Tachikawa K, Nakajima S, Ozono K. Common mutations F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur J Pediatr. 2005; 164:277–82.
Article61. Whyte MP, Valdes R Jr, Ryan LM, McAlister WH. Infantile hypophosphatasia: enzyme replacement therapy by intravenous infusion of alkaline phosphatase-rich plasma from patients with Paget bone disease. J Pediatr. 1982; 101:379–86.
Article62. Whyte MP, Greenberg CR, Salman NJ, Bober MB, McAlister WH, Wenkert D, et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012; 366:904–13.
Article63. Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016; 101:334–42.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Myositis Ossificans in Rectus Abdominis Muscle: Case Report
- Observation of tooth mineralization of Korean neonates according to gestational age
- Bilateral Transverse (Bowdler) Fibular Spurs with Hypophosphatasia in an Adolescent Girl
- Severe pulmonary mineralization in a dog with pituitary-dependent hyperadrenocorticism: a case report
- Application of Botulinum Toxin in Pain Management
