Down-Regulation of TGF-β Expression Sensitizes the Resistance of Hepatocellular Carcinoma Cells to Sorafenib
- Affiliations
-
- 1Department of Oncology, Affiliated Hospital of Yanbian University, Yanji, Jilin Province, P.R. China.
- 2Institute for Cancer Research, Yonsei University College of Medicine, Seoul, Korea.
- 3Severance Biomedical Science Institute, Yonsei University College of Medicine, Seoul, Korea.
- 4Department of Internal Medicine, Yonsei University College of Medicine, Seoul, Korea. choihj@yuhs.ac jjs109@yuhs.ac
- KMID: 2418924
- DOI: http://doi.org/10.3349/ymj.2017.58.5.899
Abstract
- PURPOSE
Sorafenib, a multikinase inhibitor, is the standard therapy for patients with advanced-stage hepatocellular carcinoma (HCC). However, resistance develops to the treatment, therefore, we tried to unravel the underlying mechanism in the resistance of HCC cells to sorafenib via the development of more effective therapeutic strategies.
MATERIALS AND METHODS
Various liver cancer cell lines were treated with either sorafenib only or with sorafenib after infection of adenovirus expressing short hairpin RNA (shRNA) against transforming growth factor-β (TGF-β) and p38 activity was examined using western blotting.
RESULTS
p38 MAP kinase activity was inhibited by low concentrations of sorafenib, which could potentially lead to sorafenib resistance in HCC cell lines. Subsequently, we used constitutive form of MKK3/6 (MKK3/6E) to confirm that massive cell death was induced by the activation of p38, and demonstrated the ability to activate p38 without any stimulation. In addition, sorafenib resistance was reduced by the activation of p38. Subsequently, we confirmed that TGF-β shRNA effectively recovered the phosphorylation of p38 inhibited by sorafenib, and increased the sensitivity of HCC cells to sorafenib, thereby inducing cell death and overcoming the resistance of HCC cells to sorafenib.
CONCLUSION
Our study provides a new therapeutic strategy for HCC that overcomes the resistance of HCC to sorafenib by down-regulation of TGF-β.
Keyword
MeSH Terms
-
Adenoviridae/metabolism
Animals
Antineoplastic Agents/pharmacology/therapeutic use
Carcinoma, Hepatocellular/*metabolism/*pathology
Cell Death/drug effects
Cell Line, Tumor
Down-Regulation/*drug effects
Drug Resistance, Neoplasm/*drug effects
Humans
Liver Neoplasms/metabolism/*pathology
Mice, Inbred BALB C
Mice, Nude
Niacinamide/*analogs & derivatives/pharmacology/therapeutic use
Phenylurea Compounds/*pharmacology/therapeutic use
Phosphorylation/drug effects
RNA, Small Interfering/metabolism
Signal Transduction/drug effects
Transforming Growth Factor beta/*metabolism
Xenograft Model Antitumor Assays
p38 Mitogen-Activated Protein Kinases/metabolism
Antineoplastic Agents
Phenylurea Compounds
RNA, Small Interfering
Transforming Growth Factor beta
Niacinamide
p38 Mitogen-Activated Protein Kinases
Figure
-

Fig. 1 Effect of sorafenib on different HCC cell lines. (A) Hep-3B, Huh7, SK-Hep-1, SNU-182, SNU-398, and SNU-449 cells were treated with sorafenib in a dose-dependent manner. After 24 h, cell viability was tested via a MTS viability assay. IC50 of each cell line is indicated in each rectangle. Error bars represent the standard error from three independent experiments. (B) HCC cell lines were treated with sorafenib at IC50 concentrations, respectively. After 24 h, the expressions of p-p38, p38, p-ERK, p-Akt, p-Src, p-p65, and GAPDH were detected by western blot analysis. (C) HCC cell lines were treated with sorafenib in a dose-dependent manner for 24 h. and incubated for additional 14 days for clonogenic assays. (D) HCC cell lines were treated with a low dose of sorafenib (2.5 µM) for 24 h, and changes in the levels of p-Akt p-p65, p-ERK, and p-p38 expression were then detected by western blot analysis. HCC, hepatocellular carcinoma.
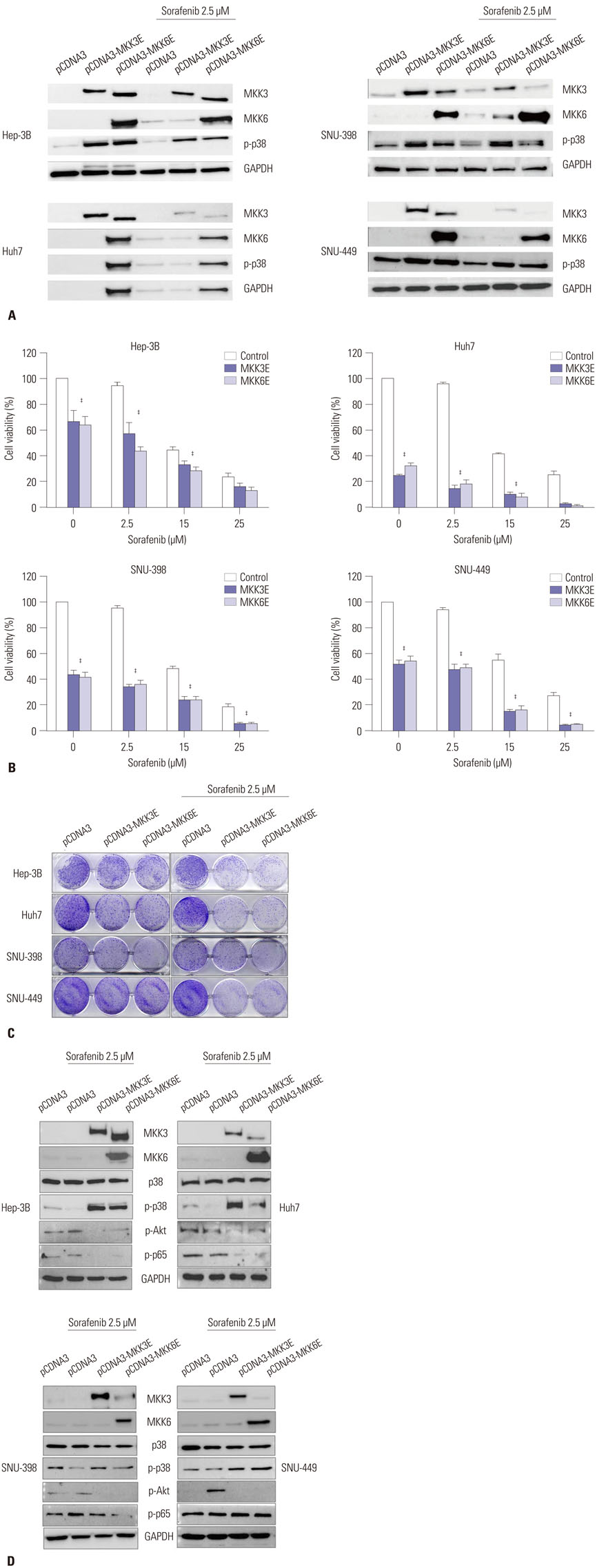
Fig. 2 MKK3/6E induced p-p38 activation and massive cell death in HCC cell lines. (A) Hep-3B, Huh7, SNU-398, and SNU-449 cells were transfected with the pCDNA3-MKK3/6E plasmid (1 µg) for 24 h, and treated with sorafenib (2.5 µM) for 24 h. Protein expressions of MKK3, MKK6, p-p38, and GAPDH were estimated via western blot analysis. (B) Hep-3B, Huh7, SNU-398, and SNU-449 cells were transfected with the pCDNA3-MKK3/6E plasmid (1 µg) for 24 h and treated with sorafenib in a dose-dependent manner for 24 h. Cell viability was examined using a MTS viability assay. Error bars represent the standard error from three independent experiments. ‡p<0.001. HCC, hepatocellular carcinoma. (C) Hep-3B, Huh7, SNU-398, and SNU-449 cells were transfected with the pCDNA3-MKK3/6E plasmid (1 µg) for 24 h, treated with sorafenib in a dose-dependent manner for 24 h, and then incubated for additional 14 days for clonogenic assays. (D) Hep-3B, Huh7, SNU-398, and SNU-449 cells were transfected with the pCDNA3-MKK3/6E plasmid (1 µg) for 24 h, and treated with sorafenib (2.5 µM) for 24 h. Protein expressions of MKK3, MKK6, p-p38, p-Akt, p-p65 and GAPDH were estimated via western blot analysis. HCC, hepatocellular carcinoma.
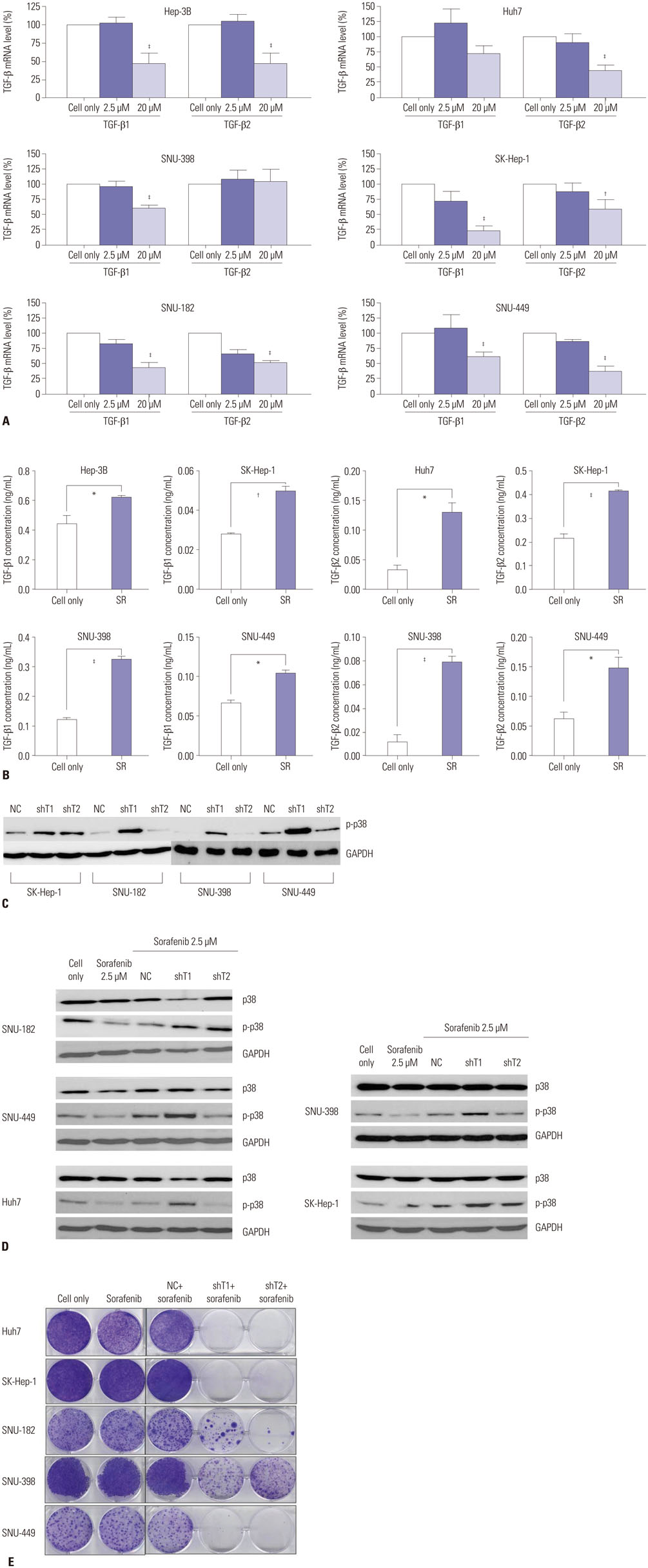
Fig. 3 Changes in TGF-β expression in response to sorafenib treatment in HCC cell lines. (A) Hep-3B, Huh7, SK-Hep-1, SNU-182, SNU-398, and SNU-449 cells were treated with sorafenib (2.5 µM, IC50) for 24 h, and TGF-β mRNA was estimated by RT-PCR. Error bars represent the standard error from three independent experiments. (B) Hep-3B, Huh7, SK-Hep-1, SNU-398, and SNU-449 cells were treated with sorafenib (2.5 µM) for 24 h and then incubated for additional 14 days for sufficient acquisition of resistance. TGF-β1/2 expression was then detected via ELISA. Error bars represent the standard error from three independent experiments. *p<0.05, †p<0.01, ‡p<0.001. TGF-β, transforming growth factor-β; HCC, hepatocellular carcinoma; RT-PCR, real-time polymerase chain reaction; ELISA, enzyme-linked immunosorbent assays; SR, sorafenib resistance. (C) SK-Hep-1, SNU-182, SNU-398, and SNU-449 cells were infected by defective adenoviruses (NC, shT1, and shT2) at 50 MOI. After 2 days, protein expression of p-p38 was detected via western blot analysis. (D) Huh7, SK-Hep-1, SNU-182, SNU-398, and SNU-449 cells were infected by defective adenoviruses (NC, shT1, and shT2) at 50 MOI. After 36 h, cells were treated with low concentration (2.5 µM) of sorafenib for 12 h. Changes in the protein expression of p38 and p-p38 were detected by western blot analysis. (E) SK-Hep-1, SNU-182, SNU-398, and SNU-449 cells were infected by defective adenoviruses (NC, shT1, and shT2) at 50 MOI. After 36 h, cells were treated with low concentration of sorafenib (2.5 µM) for 12 h, and were incubated for additional 14 days for clonogenic assays. TGF-β, transforming growth factor-β; HCC, hepatocellular carcinoma; NC, negative control; MOI, multiplicity of infection.

Fig. 4 Antitumor effects of the combined treatment of sorafenib and an adenovirus co-expressing shTGF-β and shHSP27 in BALB/c nude mice. (A) SNU-449 tumors were grown in male BALB/c nude mice. Tumors were established by subcutaneous injection of 1×107 cells and were allowed to grow to an average size of 60–100 mm3. PBS and adenoviruses were intratumorally injected every other day for a total of 3 injections. Sorafenib (30 mg/kg) was administered via gavage once daily from days 1 to 10. Tumor growth was measured every 2 days for more than 19 days using calipers. (B) Survival rates were calculated every 2 days for more than 19 days. TGF-β, transforming growth factor-β; NC, negative control; PBS, phosphate buffered saline.
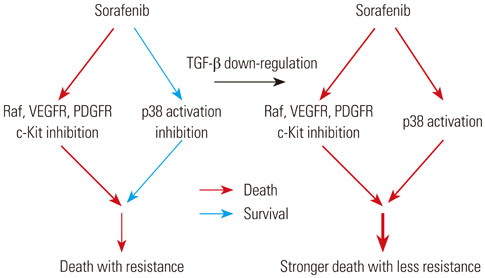
Fig. 5 A schematic diagram of sorafenib drug resistance by p38 activation inhibition and TGF-β down-regulation-induced sensitization of the resistance to sorafenib. TGF-β, transforming growth factor-β; VEGFR, vascular endothelial growth factor receptor; PDGFR, platelet-derived growth factor receptor.
Reference
-
1. McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011; 15:223–243.
Article2. Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003; 362:1907–1917.
Article3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359:378–390.
Article4. Bruix J, Sherman M. Practice Guidelines Committee, American Association for the Study of Liver Diseases. Management of hepatocellular carcinoma. Hepatology. 2005; 42:1208–1236.
Article5. Zhai B, Hu F, Jiang X, Xu J, Zhao D, Liu B, et al. Inhibition of Akt reverses the acquired resistance to sorafenib by switching protective autophagy to autophagic cell death in hepatocellular carcinoma. Mol Cancer Ther. 2014; 13:1589–1598.
Article6. Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Mol Cancer Ther. 2008; 7:3129–3140.
Article7. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004; 64:7099–7109.
Article8. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10:25–34.
Article9. Zhai B, Sun XY. Mechanisms of resistance to sorafenib and the corresponding strategies in hepatocellular carcinoma. World J Hepatol. 2013; 5:345–352.
Article10. Berasain C. Hepatocellular carcinoma and sorafenib: too many resistance mechanisms? Gut. 2013; 62:1674–1675.
Article11. Zhu YJ, Zheng B, Wang HY, Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017; 38:614–622.
Article12. Iyoda K, Sasaki Y, Horimoto M, Toyama T, Yakushijin T, Sakakibara M, et al. Involvement of the p38 mitogen-activated protein kinase cascade in hepatocellular carcinoma. Cancer. 2003; 97:3017–3026.
Article13. Massagué J. TGFbeta in cancer. Cell. 2008; 134:215–230.14. Nagaraj NS, Datta PK. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin Investig Drugs. 2010; 19:77–91.
Article15. Papageorgis P. TGFβ Signaling in Tumor Initiation, Epithelial-to-Mesenchymal Transition, and Metastasis. J Oncol. 2015; 2015:587193.16. Lin TH, Shao YY, Chan SY, Huang CY, Hsu CH, Cheng AL. High serum transforming growth factor-β1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res. 2015; 21:3678–3684.
Article17. Dropmann A, Dediulia T, Breitkopf-Heinlein K, Korhonen H, Janicot M, Weber SN, et al. TGF-β1 and TGF-β2 abundance in liver diseases of mice and men. Oncotarget. 2016; 7:19499–19518.
Article18. Wilhelm S, Carter C, Lynch M, Lowinger T, Dumas J, Smith RA, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006; 5:835–844.
Article19. Namboodiri HV, Bukhtiyarova M, Ramcharan J, Karpusas M, Lee Y, Springman EB. Analysis of imatinib and sorafenib binding to p38alpha compared with c-Abl and b-Raf provides structural insights for understanding the selectivity of inhibitors targeting the DFG-out form of protein kinases. Biochemistry. 2010; 49:3611–3618.
Article20. Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006; 2:358–364.
Article21. Grossi V, Liuzzi M, Murzilli S, Martelli N, Napoli A, Ingravallo G, et al. Sorafenib inhibits p38α activity in colorectal cancer cells and synergizes with the DFG-in inhibitor SB202190 to increase apoptotic response. Cancer Biol Ther. 2012; 13:1471–1481.
Article22. Edelstein ML, Abedi MR, Wixon J. Gene therapy clinical trials worldwide to 2007--an update. J Gene Med. 2007; 9:833–842.
Article23. Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006; 407:597–612.
Article24. Yang Y, Shen Y, Liu H, Yao X. Molecular dynamics simulation and free energy calculation studies of the binding mechanism of allosteric inhibitors with p38α MAP kinase. J Chem Inf Model. 2011; 51:3235–3246.
Article25. Raingeaud J, Whitmarsh AJ, Barrett T, Dérijard B, Davis RJ. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol. 1996; 16:1247–1255.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Glutamine synthetase mediates sorafenib sensitivity in β-catenin-active hepatocellular carcinoma cells
- Vimentin as a potential therapeutic target in sorafenib resistant HepG2, a HCC model cell line
- C-terminal-truncated HBV X promotes hepato-oncogenesis through inhibition of tumor-suppressive β-catenin/BAMBI signaling
- Treatments Other than Sorafenib for Patients with Advanced Hepatocellular Carcinoma
- Treatment options after sorafenib failure in patients with hepatocellular carcinoma
