Novel ATP8B1 Gene Mutations in a Child with Progressive Familial Intrahepatic Cholestasis Type 1
- Affiliations
-
- 1Department of Pediatrics, Asan Medical Center Children's Hospital, University of Ulsan College of Medicine, Seoul, Korea. seakhee.oh@amc.seoul.kr
- KMID: 2457750
- DOI: http://doi.org/10.5223/pghn.2019.22.5.479
Abstract
- Progressive familial intrahepatic cholestasis (PFIC) is a group of severe genetic disorders, inherited in an autosomal recessive manner, causing cholestasis of hepatocellular origin, later progressing to biliary cirrhosis and liver failure. This is the first report of PFIC type 1 with novel compound heterozygous mutations in Korea. The patient was presented with intrahepatic cholestasis, a normal level of serum γ-glutamyl transferase, steatorrhea, and growth failure. Genetic testing of this patient revealed novel compound heterozygous mutations (p.Glu585Ter and p.Leu749Pro) in the ATP8B1 gene. After a liver transplantation at age 19 months, the patient developed severe post-transplant steatohepatitis.
Keyword
MeSH Terms
Figure
-
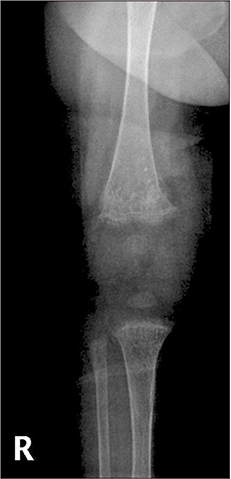
Fig. 1 Rickets and osteoporosis of right knee in the patient.
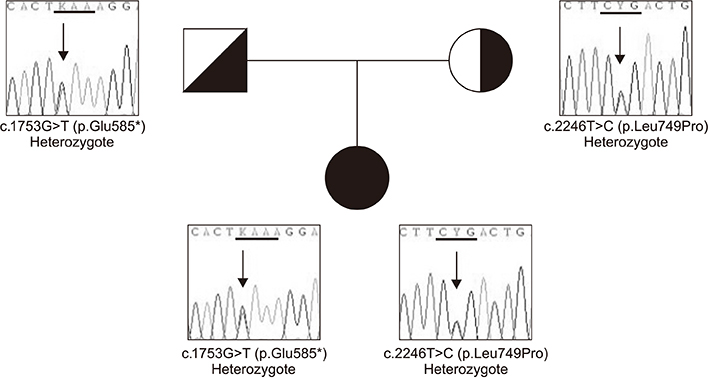
Fig. 2 Variants of the ATP8B1 gene in the patient. The patient inherited a missense mutation from her mother (L749P) and a nonsense mutation from her father (E585X).
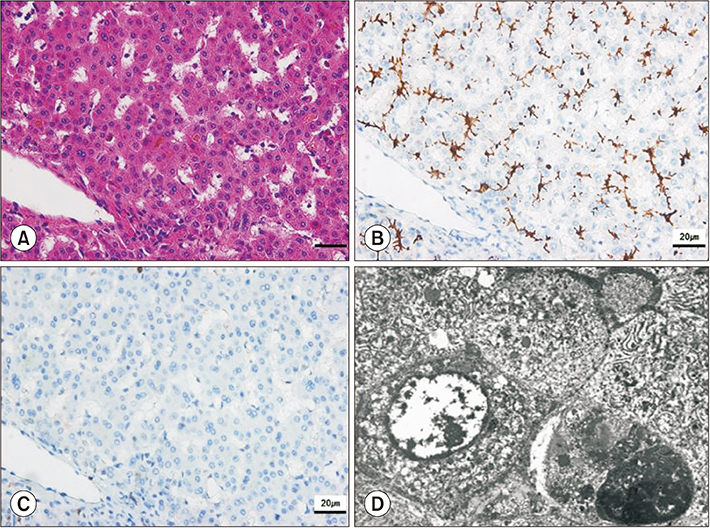
Fig. 3 Biopsy of the explanted liver. (A) Mild portal inflammation with focal bile duct absence, periportal and perisinusoidal fibrosis, and moderate cholestasis (H&E, ×400). (B) Bile salt export pump (BSEP) protein was normally distributed on immunohistochemistry (IHC) staining of the patient's liver (IHC with anti-BSEP) (H&E, ×400). (C) Decreased CD10 staining suggested fewer expressed canaliculi (IHC with CD10) (H&E, ×400). (D) Dilated canaliculus with coarse and granular bile and intraductal bile plugs (electron microscopy) (H&E, ×4,000).
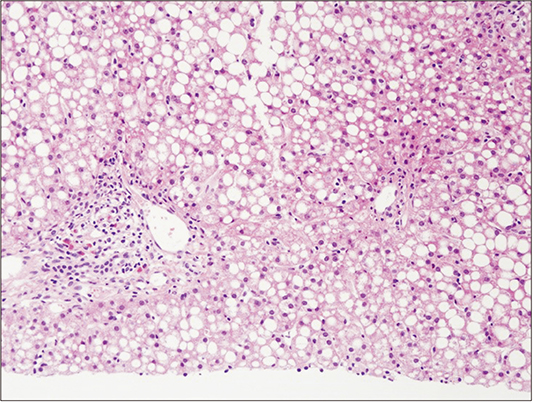
Fig. 4 Liver biopsy of the patient 4 months after transplantation. The biopsy shows macrovesicular steatosis with focal minimal lobular activity (H&E, ×200).
Cited by 1 articles
-
Liver transplantation in pediatric patients with progressive familial intrahepatic cholestasis: Single center experience of seven cases
Jung-Man Namgoong, Shin Hwang, Hyunhee Kwon, Suhyeon Ha, Kyung Mo Kim, Seak Hee Oh, Seung-Mo Hong
Ann Hepatobiliary Pancreat Surg. 2022;26(1):69-75. doi: 10.14701/ahbps.21-114.
Reference
-
1. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009; 4:1.
Article2. Cavestro GM, Frulloni L, Cerati E, Ribeiro LA, Corrente V, Sianesi M, et al. Progressive familial intrahepatic cholestasis. Acta Biomed. 2002; 73:53–56.3. Amer S, Hajira A. A comprehensive review of progressive familial intrahepatic cholestasis (PFIC): genetic disorders of hepatocanalicular transporters. Gastroenterology Res. 2014; 7:39–43.
Article4. Srivastava A. Progressive familial intrahepatic cholestasis. J Clin Exp Hepatol. 2014; 4:25–36.
Article5. Gaur K, Sakhuja P. Progressive familial intrahepatic cholestasis: a comprehensive review of a challenging liver disease. Indian J Pathol Microbiol. 2017; 60:2–7.6. Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010; 51:1645–1655.
Article7. Arnell H, Nemeth A, Annerén G, Dahl N. Progressive familial intrahepatic cholestasis (PFIC): evidence for genetic heterogeneity by exclusion of linkage to chromosome 18q21-q22. Hum Genet. 1997; 100:378–381.
Article8. Chen HL, Chang PS, Hsu HC, Ni YH, Hsu HY, Lee JH, et al. FIC1 and BSEP defects in Taiwanese patients with chronic intrahepatic cholestasis with low gamma-glutamyltranspeptidase levels. J Pediatr. 2002; 140:119–124.
Article9. Hori T, Egawa H, Takada Y, Ueda M, Oike F, Ogura Y, et al. Progressive familial intrahepatic cholestasis: a single-center experience of living-donor liver transplantation during two decades in Japan. Clin Transplant. 2011; 25:776–785.
Article10. Numakura C, Abukawa D, Kimura T, Tanabe S, Hayasaka K. A case of progressive familial intrahepatic cholestasis type 1 with compound heterozygous mutations of ATP8B1. Pediatr Int. 2011; 53:107–110.
Article11. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424.
Article12. Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003; 39:447–452.
Article13. Luketic VA, Shiffman ML. Benign recurrent intrahepatic cholestasis. Clin Liver Dis. 1999; 3:509–528. viii
Article14. Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004; 40:27–38.
Article15. Paulusma CC, Elferink RP, Jansen PL. Progressive familial intrahepatic cholestasis type 1. Semin Liver Dis. 2010; 30:117–124.
Article16. Maillette de Buy Wenniger L, Beuers U. Bile salts and cholestasis. Dig Liver Dis. 2010; 42:409–418.
Article17. Sharma A, Poddar U, Agnihotry S, Aggarwal R. A novel truncation mutation in ATP8B1 gene in progressive familial intrahepatic cholestasis. Indian Pediatr. 2016; 53:1099–1101.18. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998; 18:219–224.
Article19. Deng BC, Lv S, Cui W, Zhao R, Lu X, Wu J, et al. Novel ATP8B1 mutation in an adult male with progressive familial intrahepatic cholestasis. World J Gastroenterol. 2012; 18:6504–6509.
Article20. Müllenbach R, Bennett A, Tetlow N, Patel N, Hamilton G, Cheng F, et al. ATP8B1 mutations in British cases with intrahepatic cholestasis of pregnancy. Gut. 2005; 54:829–834.
Article21. Alvarez L, Jara P, Sánchez-Sabaté E, Hierro L, Larrauri J, Díaz MC, et al. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004; 13:2451–2460.
Article22. Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997; 25:519–523.
Article23. Aydogdu S, Cakir M, Arikan C, Tumgor G, Yuksekkaya HA, Yilmaz F, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007; 11:634–640.
Article24. Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawa M, Tanaka K. Intractable diarrhea after liver transplantation for Byler's disease: successful treatment with bile adsorptive resin. Liver Transpl. 2002; 8:714–716.
Article25. Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011; 31:3–10.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Phenotypic and Molecular Characteristics of Children with Progressive Familial Intrahepatic Cholestasis in South China
- Benign Recurrent Intrahepatic Cholestasis with a Single Heterozygote Mutation in the ATP8B1 Gene
- Presentation of Progressive Familial Intrahepatic Cholestasis Type 3 Mimicking Wilson Disease: Molecular Genetic Diagnosis and Response to Treatment
- Progressive Familial Intrahepatic Cholestasis in Korea: A Clinicopathological Study of Five Patients
- Successful Use of Bortezomib for Recurrent Progressive Familial Intrahepatic Cholestasis Type II After Liver Transplantation: A Pediatric Case with a 9-Year Follow-Up
