Progressive Familial Intrahepatic Cholestasis in Korea: A Clinicopathological Study of Five Patients
- Affiliations
-
- 1Department of Pathology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea. esyu@amc.seoul.kr
- 2Department of Pathology, Soonchunhyang University Cheonan Hospital, Cheonan, Korea.
- 3Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 4Medical Genetics Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 5Asan Liver Center, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- KMID: 2454605
- DOI: http://doi.org/10.4132/jptm.2019.05.03
Abstract
- BACKGROUND
Progressive familial intrahepatic cholestasis (PFIC) is a heterogeneous group of autosomal recessive liver diseases that present as neonatal cholestasis. Little is known of this disease in Korea.
METHODS
The records of five patients histologically diagnosed with PFIC, one with PFIC1 and four with PFIC2, by liver biopsy or transplant were reviewed, and ATP8B1 and ABCB11 mutation status was analyzed by direct DNA sequencing. Clinicopathological characteristics were correlated with genetic mutations.
RESULTS
The first symptom in all patients was jaundice. Histologically, lobular cholestasis with bile plugs was the main finding in all patients, whereas diffuse or periportal cholestasis was identified only in patients with PFIC2. Giant cells and ballooning of hepatocytes were observed in three and three patients with PFIC2, respectively, but not in the patient with PFIC1. Immunostaining showed total loss of bile salt export pump in two patients with PFIC2 and focal loss in two. Lobular and portal based fibrosis were more advanced in PFIC2 than in PFIC1. ATP8B1 and ABCB11 mutations were identified in one PFIC1 and two PFIC2 patients, respectively. One PFIC1 and three PFIC2 patients underwent liver transplantation (LT). At age 7 months, one PFIC2 patient was diagnosed with concurrent hepatocellular carcinoma and infantile hemangioma in an explanted liver. The patient with PFIC1 developed steatohepatitis after LT. One patient showed recurrence of PFIC2 after 10 years and underwent LT.
CONCLUSIONS
PFIC is not rare in patients with neonatal cholestasis of unknown origin. Proper clinicopathologic correlation and genetic testing can enable early detection and management.
MeSH Terms
Figure
-

Fig. 1. Histologic findings in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Bland canalicular cholestasis and small cell change of hepatocytes with lobular disarray in our PFIC-1 patient. Bile duct proliferation with cholangiolar cholestasis (B), ballooning change (C), and giant cell transformation of hepatocytes (D) in patients with PFIC-2.

Fig. 2. Stages of fibrosis in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Cirrhosis with diffuse lobular fibrosis in a PFIC2 patient, as shown by Masson trichrome staining. (B) Periportal fibrosis with mild lobular fibrosis in the one patient with PFIC1.
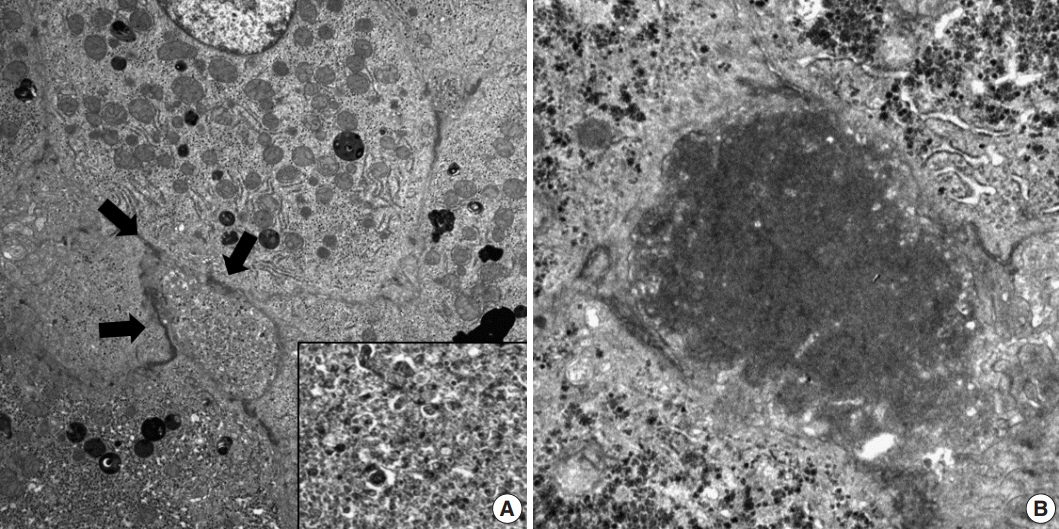
Fig. 3. Electron microscopic findings in patients with progressive familial intrahepatic cholestasis (PFIC). (A) Dilated canaliculi (arrows) with coarse granular bile in the PFIC1 patient. (B) Amorphous and dense bile in PFIC2 patients.
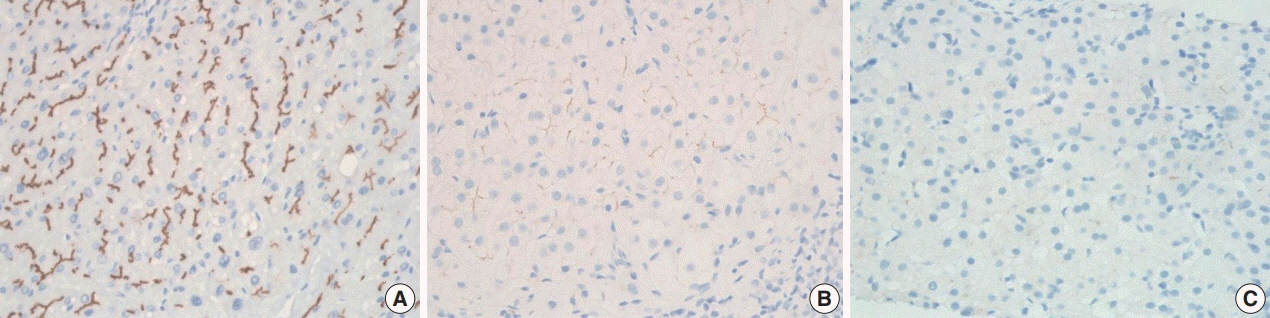
Fig. 4. Immunohistochemical staining for bile salt export pump: normal canalicular expression (A), focal loss (B), and total loss (C).
Cited by 1 articles
-
Liver transplantation in pediatric patients with progressive familial intrahepatic cholestasis: Single center experience of seven cases
Jung-Man Namgoong, Shin Hwang, Hyunhee Kwon, Suhyeon Ha, Kyung Mo Kim, Seak Hee Oh, Seung-Mo Hong
Ann Hepatobiliary Pancreat Surg. 2022;26(1):69-75. doi: 10.14701/ahbps.21-114.
Reference
-
1. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009; 4:1.
Article2. Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012; 36 Suppl 1:S26–35.
Article3. Clayton RJ, Iber FL, Ruebner BH, McKusick VA. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. Am J Dis Child. 1969; 117:112–24.4. Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a Ptype ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998; 18:219–24.
Article5. Strautnieks SS, Bull LN, Knisely AS, et al. A gene encoding a liverspecific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998; 20:233–8.
Article6. van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001; 21:535–44.
Article7. Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH. Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol. 2010; 52:258–71.
Article8. Park YN, Kim HG, Chon CY, et al. Histological grading and staging of chronic hepatitis standardized guideline proposed by the Korean Study Group for the Pathology of Digestive Diseases. Korean J Pathol. 1999; 33:337–46.9. Yu E, Korean Study Group for the Pathology of Digestive Diseases. Histologic grading and staging of chronic hepatitis: on the basis of standardized guideline proposed by the Korean Study Group for the Pathology of Digestive Diseases. Taehan Kan Hakhoe Chi. 2003; 9:42–6.10. Nemesánszky E, Lott JA. Gamma-glutamyltransferase and its isoenzymes: progress and problems. Clin Chem. 1985; 31:797–803.
Article11. Oude Elferink RP, Paulusma CC, Groen AK. Hepatocanalicular transport defects: pathophysiologic mechanisms of rare diseases. Gastroenterology. 2006; 130:908–25.
Article12. Evason K, Bove KE, Finegold MJ, et al. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol. 2011; 35:687–96.13. Alissa FT, Jaffe R, Shneider BL. Update on progressive familial intrahepatic cholestasis. J Pediatr Gastroenterol Nutr. 2008; 46:241–52.
Article14. Morotti RA, Suchy FJ, Magid MS. Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis. 2011; 31:3–10.
Article15. Hori T, Nguyen JH, Uemoto S. Progressive familial intrahepatic cholestasis. Hepatobiliary Pancreat Dis Int. 2010; 9:570–8.16. Bull LN, Carlton VE, Stricker NL, et al. Genetic and morphological findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC-1] and Byler syndrome): evidence for heterogeneity. Hepatology. 1997; 26:155–64.
Article17. Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: progressive familial intrahepatic cholestasis type 1. Eur J Hum Genet. 2013; Aug. 28. [Epub]. https://doi.org/10.1038/ejhg.2013.186.
Article18. Gonzales E, Spraul A, Jacquemin E. Clinical utility gene card for: progressive familial intrahepatic cholestasis type 2. Eur J Hum Genet. 2013; Aug. 28. [Epub]. https://doi.org/10.1038/ejhg.2013.187.
Article19. Klomp LW, Vargas JC, van Mil SW, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004; 40:27–38.20. Liu LY, Wang XH, Wang ZL, Zhu QR, Wang JS. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT. J Pediatr Gastroenterol Nutr. 2010; 50:179–83.21. Jacquemin E, Hermans D, Myara A, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997; 25:519–23.
Article22. Amer S, Hajira A. A comprehensive review of progressive familial intrahepatic cholestasis (PFIC): genetic disorders of hepatocanalicular transporters. Gastroenterology Res. 2014; 7:39–43.
Article23. Bustorff-Silva J, Sbraggia Neto L, Olímpio H, et al. Partial internal biliary diversion through a cholecystojejunocolonic anastomosis: a novel surgical approach for patients with progressive familial intrahepatic cholestasis: a preliminary report. J Pediatr Surg. 2007; 42:1337–40.24. Aydogdu S, Cakir M, Arikan C, et al. Liver transplantation for progressive familial intrahepatic cholestasis: clinical and histopathological findings, outcome and impact on growth. Pediatr Transplant. 2007; 11:634–40.
Article25. Hori T, Egawa H, Takada Y, et al. Progressive familial intrahepatic cholestasis: a single-center experience of living-donor liver transplantation during two decades in Japan. Clin Transplant. 2011; 25:776–85.
Article26. Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007; 84:1361–3.
Article27. Lykavieris P, van Mil S, Cresteil D, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003; 39:447–52.
Article28. Miyagawa-Hayashino A, Egawa H, Yorifuji T, et al. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl. 2009; 15:610–8.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Presentation of Progressive Familial Intrahepatic Cholestasis Type 3 Mimicking Wilson Disease: Molecular Genetic Diagnosis and Response to Treatment
- Novel ATP8B1 Gene Mutations in a Child with Progressive Familial Intrahepatic Cholestasis Type 1
- Familial Benign Recurrent Intrahepatic Cholestasis
- Hepatocellular carcinoma associated with progressive intrahepatic familial cholestasis type 2: a case report
- Early Diagnosis of ABCB11 Spectrum Liver Disorders by Next Generation Sequencing
