Presentation of Progressive Familial Intrahepatic Cholestasis Type 3 Mimicking Wilson Disease: Molecular Genetic Diagnosis and Response to Treatment
- Affiliations
-
- 1Division of Digestive Diseases and Section of Transplantation and Immunology, Department of Medicine and Surgery, Yale University School of Medicine, New Haven, CT, USA. salih.boga@yale.edu
- 2Department of Pathology, Yale University School of Medicine, New Haven, CT, USA.
- KMID: 2068799
- DOI: http://doi.org/10.5223/pghn.2015.18.3.202
Abstract
- Progressive familial intrahepatic cholestasis type 3 (PFIC3) is an autosomal recessive disorder of cholestasis of hepatocellular origin, typically seen in infancy or childhood caused by a defect in the ABCB4 located on chromosome 7. Here we report on an older patient, aged 15, who presented with biochemical testing that led to an initial consideration of a diagnosis of Wilson disease (WD) resulting in a delayed diagnosis of PFIC3. Diagnosis of PFIC3 was later confirmed by molecular studies that identified novel mutations in the ABCB4 gene. Cholestasis due to PFIC3 can cause elevated hepatic copper and increased urine copper excretion that overlap with current diagnostic criteria for WD. Molecular diagnostics are very useful for establishing the diagnosis of PFIC3. Ursodeoxycholic acid ameliorates cholestasis in PFIC3, and may help mediate a reduction in hepatic copper content in response to treatment.
MeSH Terms
Figure
-
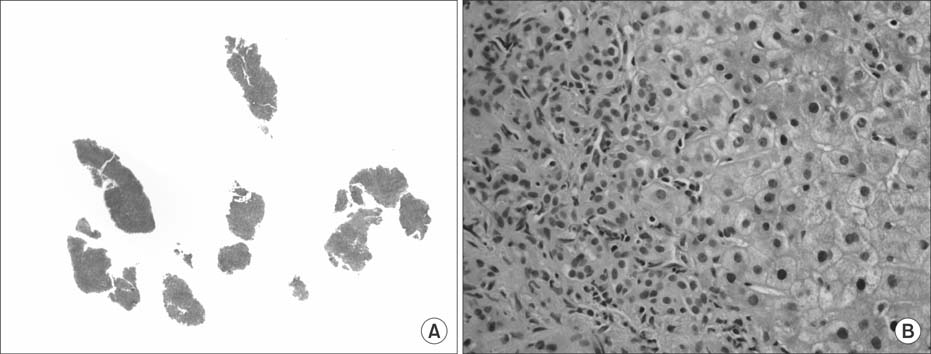
Fig. 1 (A) Low magnification of the original biopsy which was extensively fragmentated and showed areas of bridging fibrosis (H&E, ×40). (B) Higher magnification showing ductular reaction, mild chronic inflammation and neutrophilic pericholangitis (H&E, ×200).
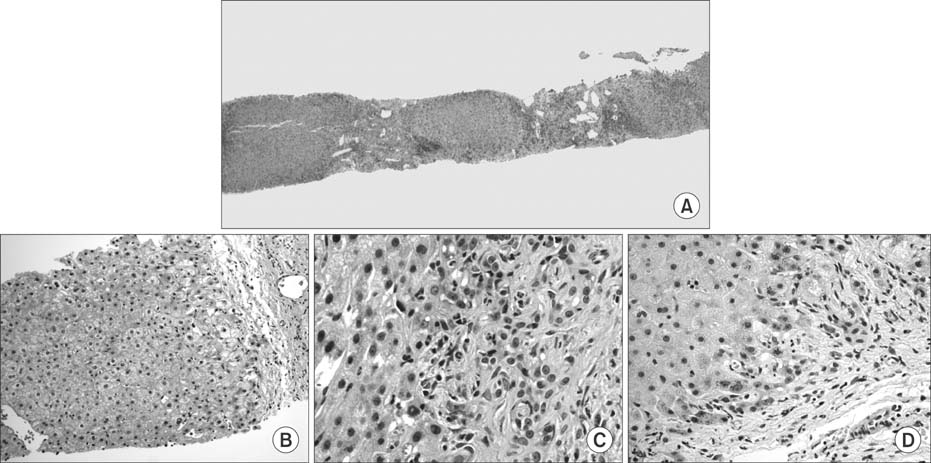
Fig. 2 (A) Low magnification of the follow-up biopsy showing cirrhosis (H&E, ×100). (B) Pale staining of periseptal hepatocyte suggestive of cholate stasis was subtle and focal (H&E, ×200). (C) Higher magnifcation showing ducutular reaction, chronic inflammation and neutrophilic pericholangitis. Brownish granular pigment representing copper can also be seen (H&E ×400). (D) Copper accumulation in the periseptal hepatocytes highlighted as red granules (rhodanine stain, ×400).
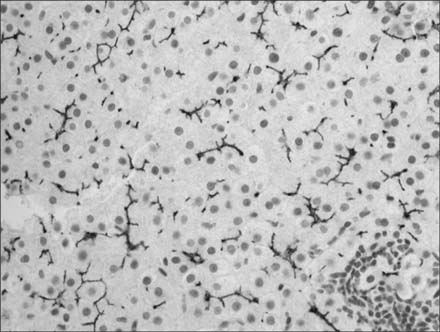
Fig. 3 Immunostaining for class III multidrug resistance shows normal canalicular staining (×200).
Reference
-
1. Jacquemin E. Progressive familial intrahepatic cholestasis. Genetic basis and treatment. Clin Liver Dis. 2000; 4:753–763.2. Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001; 21:551–562.
Article3. Baussan C, Cresteil D, Gonzales E, Raynaud N, Dumont M, Bernard O, et al. Genetic cholestatic liver diseases: the example of progressive familial intrahepatic cholestasis and related disorders. Acta Gastroenterol Belg. 2004; 67:179–183.4. Shneider BL. ABCB4 disease presenting with cirrhosis and copper overload-potential confusion with Wilson disease. J Clin Exp Hepatol. 2011; 1:115–117.
Article5. Ramraj R, Finegold MJ, Karpen SJ. Progressive familial intrahepatic cholestasis type 3: overlapping presentation with Wilson disease. Clin Pediatr (Phila). 2012; 51:689–691.
Article6. European Association for Study of Liver. EASL Clinical Practice Guidelines: Wilson's disease. J Hepatol. 2012; 56:671–685.7. Sternlieb I, Quintana N. Biliary proteins and ductular ultrastructure. Hepatology. 1985; 5:139–143.
Article8. Wendum D, Barbu V, Rosmorduc O, Arrivé L, Fléjou JF, Poupon R. Aspects of liver pathology in adult patients with MDR3/ABCB4 gene mutations. Virchows Arch. 2012; 460:291–298.
Article9. Sannier A, Ganne N, Tepper M, Ziol M. MDR3 immunostaining on frozen liver biopsy samples is not a sensitive diagnostic tool for the detection of heterozygous MDR3/ABCB4 gene mutations. Virchows Arch. 2012; 460:535–537.
Article10. Jacquemin E, Hermans D, Myara A, Habes D, Debray D, Hadchouel M, et al. Ursodeoxycholic acid therapy in pediatric patients with progressive familial intrahepatic cholestasis. Hepatology. 1997; 25:519–523.
Article11. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009; 4:1.
Article12. Lai J, Taouli B, Iyer KR, Arnon R, Thung SN, Magid MS, et al. Cholangiolocellular carcinoma in a pediatric patient with small duct sclerosing cholangitis: a case report. Semin Liver Dis. 2012; 32:360–366.
Article13. Jacquemin E, De Vree JM, Cresteil D, Sokal EM, Sturm E, Dumont M, et al. The wide spectrum of multidrug resistance 3 deficiency: from neonatal cholestasis to cirrhosis of adulthood. Gastroenterology. 2001; 120:1448–1458.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Novel ATP8B1 Gene Mutations in a Child with Progressive Familial Intrahepatic Cholestasis Type 1
- The etiologies of neonatal cholestasis
- Early Diagnosis of ABCB11 Spectrum Liver Disorders by Next Generation Sequencing
- The First Korean Adult Case of Progressive Familial Intrahepatic Cholestasis Type 7 with Novel USP53 Splicing Variants by Next Generation Sequencing
- Hepatocellular carcinoma associated with progressive intrahepatic familial cholestasis type 2: a case report
