Pediatr Gastroenterol Hepatol Nutr.
2012 Jun;15(2):122-126.
Benign Recurrent Intrahepatic Cholestasis with a Single Heterozygote Mutation in the ATP8B1 Gene
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. i101016@skku.edu
- 2Department of Laboratory Medicine & Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
Abstract
- Benign recurrent intrahepatic cholestasis (BRIC) is a rare autosomal recessive inherited disorder characterized by multiple recurrent episodes of severe cholestatic jaundice without obstruction of extrahepatic bile duct. We present the case of a 7-year-old boy with BRIC confirmed by mutation analysis in the ATP8B1 gene and typical clinical manifestation. Despite inheritance of BRIC, we detected a mutation on only one allele. To our knowledge, this is the first report of BRIC with a confirmed single heterozygote novel mutation in the ATP8B1 gene in Korea.
MeSH Terms
Figure
-

Fig. 1 Direct sequencing of PCR products amplified from exon 22 of the ATP8B1 gene. The patient displayed a heterozygote frame shift mutation by insertion of a nucleotide C at 2610_2611.

Fig. 2 Growth parameters with height and body weight. From the third attack (2007) to date, catch-up growth is not shown.
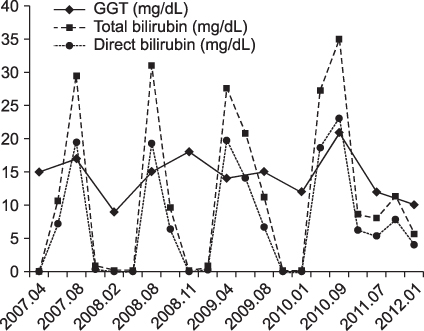
Fig. 3 The total bilirubin, direct bilirubin and GGT level changes over the patient's natural history. Between attacks, the patient was in complete remission with no cholestasis. GGT was normal during an episode with profound cholestasis.
Reference
-
1. Folvik G, Hilde O, Helge GO. Benign recurrent intrahepatic cholestasis: review and long-term follow-up of five cases. Scand J Gastroenterol. 2012. 47:482–488.
Article2. Kim OY, Sung BY, Kowg GD, Yoon HS, Shin YM, Oh HT, et al. A case of nonfamilial benign recurrent intrahepatic cholestasis. Korean J Hepatol. 1998. 4:188–193.3. Ko JS, Seo JK. The etiologies of neonatal cholestasis. Korean J Pediatr. 2007. 50:835–840.
Article4. Klomp LW, Vargas JC, van Mil SW, Pawlikowska L, Strautnieks SS, van Eijk MJ, et al. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 2004. 40:27–38.
Article5. van der Woerd WL, van Mil SW, Stapelbroek JM, Klomp LW, van de Graaf SF, Houwen RH. Familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract Res Clin Gastroenterol. 2010. 24:541–553.
Article6. Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003. 39:447–452.
Article7. Summerskill WH, Walshe JM. Benign recurrent intrahepatic "obstructive" jaundice. Lancet. 1959. 2:686–690.8. Tygstrup N, Jensen B. Intermittent intrahepatic cholestasis of unknown etiology in five young males from the Faroe Islands. Acta Med Scand. 1969. 185:523–530.
Article9. Houwen RH, Baharloo S, Blankenship K, Raeymaekers P, Juyn J, Sandkuijl LA, et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet. 1994. 8:380–386.
Article10. Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998. 18:219–224.
Article11. Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH. Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol. 2010. 52:258–271.
Article12. Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009. 4:1.
Article13. Liu LY, Wang XH, Wang ZL, Zhu QR, Wang JS. Characterization of ATP8B1 gene mutations and a hot-linked mutation found in Chinese children with progressive intrahepatic cholestasis and low GGT. J Pediatr Gastroenterol Nutr. 2010. 50:179–183.
Article14. Bull LN, Juijn JA, Liao M, van Eijk MJ, Sinke RJ, Stricker NL, et al. Fine-resolution mapping by haplotype evaluation: the examples of PFIC1 and BRIC. Hum Genet. 1999. 104:241–248.
Article15. Tygstrup N, Steig BA, Juijn JA, Bull LN, Houwen RH. Recurrent familial intrahepatic cholestasis in the Faeroe Islands. Phenotypic heterogeneity but genetic homogeneity. Hepatology. 1999. 29:506–508.
Article16. Mezey E, Burns C, Burdick JF, Braine HG. A case of severe benign intrahepatic cholestasis treated with liver transplantation. Am J Gastroenterol. 2002. 97:475–477.
Article17. van Mil SW, Klomp LW, Bull LN, Houwen RH. FIC1 disease: a spectrum of intrahepatic cholestatic disorders. Semin Liver Dis. 2001. 21:535–544.
Article18. van Ooteghem NA, Klomp LW, van Berge-Henegouwen GP, Houwen RH. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J Hepatol. 2002. 36:439–443.
Article19. van Mil SW, van der Woerd WL, van der Brugge G, Sturm E, Jansen PL, Bull LN, et al. Benign recurrent intrahepatic cholestasis type 2 is caused by mutations in ABCB11. Gastroenterology. 2004. 127:379–384.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A case of benign recurrent intrahepatic cholestasis
- Novel ATP8B1 Gene Mutations in a Child with Progressive Familial Intrahepatic Cholestasis Type 1
- Familial Benign Recurrent Intrahepatic Cholestasis
- A Case of Nonfamilial Benign Recurrent Intrahepatic Cholestasis
- Phenotypic and Molecular Characteristics of Children with Progressive Familial Intrahepatic Cholestasis in South China
