Ferroptosis: A Novel Anti-tumor Action for Cisplatin
- Affiliations
-
- 1Cancer Center, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China. xhzlwg@163.com, lizhy@hotmail.com
- 2Cancer center, Xianning Center Hospital, Xianning, China.
- KMID: 2411134
- DOI: http://doi.org/10.4143/crt.2016.572
Abstract
- PURPOSE
Ferroptosis is a new mode of regulated cell death, which is completely distinct from other cell death modes based on morphological, biochemical, and genetic criteria. This study evaluated the therapeutic role of ferroptosis in classic chemotherapy drugs, including the underlying mechanism.
MATERIALS AND METHODS
Cell viabilitywas detected by using the methylthiazoltetrazlium dye uptake method. RNAiwas used to knockout iron-responsive element binding protein 2, and polymerase chain reaction, western blot was used to evaluate the efficiency. Intracellular reduced glutathione level and glutathione peroxidases activitywere determined by related assay kit. Intracellularreactive oxygen species levelswere determined by flowcytometry. Electron microscopywas used to observe ultrastructure changes in cell.
RESULTS
Among five chemotherapeutic drugs screened in this study, cisplatin was found to be an inducer for both ferroptosis and apoptosis in A549 and HCT116 cells. The depletion of reduced glutathione caused by cisplatin and the inactivation of glutathione peroxidase played the vital role in the underlying mechanism. Besides, combination therapy of cisplatin and erastin showed significant synergistic effect on their anti-tumor activity.
CONCLUSION
Ferroptosis had great potential to become a new approach in anti-tumor therapies and make up for some classic drugs, which open up a new way for their utility in clinic.
Keyword
MeSH Terms
Figure
-
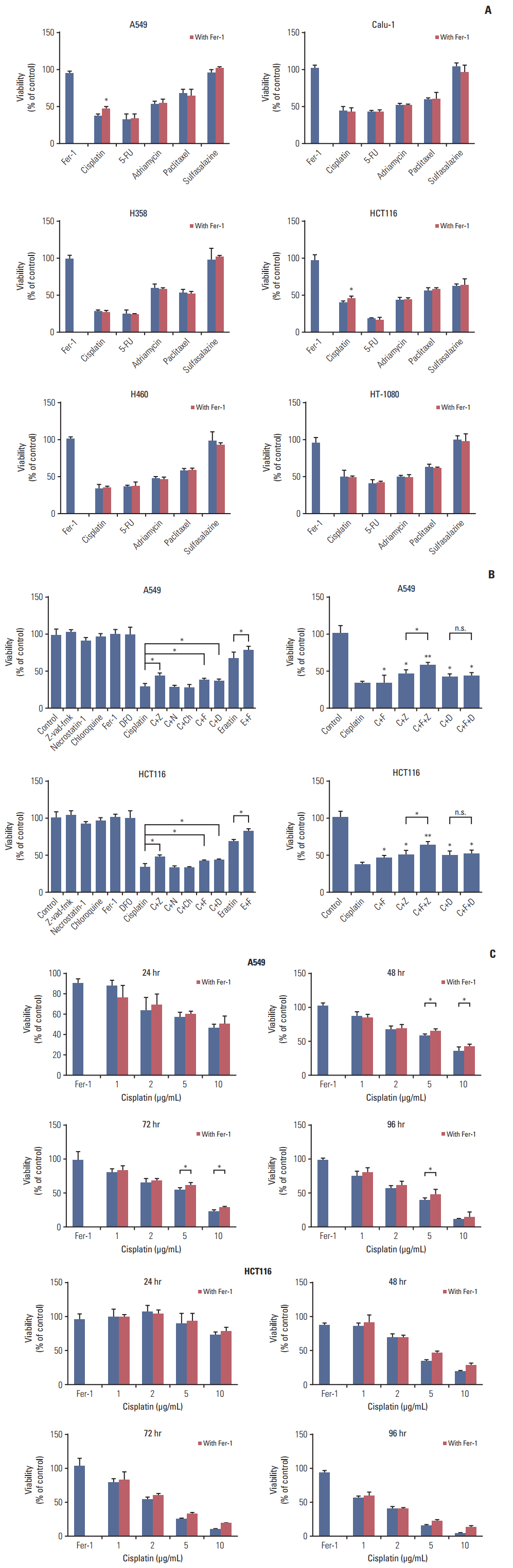
Fig. 1. Cisplatin induced both apoptosis and ferroptosis on A549 and HCT116 cells. (A) A549, NCIH358, NCIH460, Calu-1, HCT116, and HT-1080 cells received treatments of cisplatin (5 μg/mL), fluorouracil (5-FU, 20 μg/mL), adriamycin (10 μg/mL), paclitaxel (10 μmol/L), and sulfasalazine (0.8 mmol/L), respectively in the absence or presence of ferrostatin-1 (Fer-1, 0.5 μmol/L) for 48 hours. (B) A549 and HCT116 cells were under the treatment of cisplatin (5 μg/mL) for 48 hours with different cell death inhibitors as described. C, cisplatin; Z, z-vad-fmk (20 μmol/L); N, necrostatin-1 (30 μmol/L); Ch, chloroquine (5 μg/mL); F, ferrostatin-1 (Fer-1, 0.5 μmol/L); D, deferoxamine, DFO (50 μmol/L); E, erastin (10 μmol/L); n.s., not significant. (C) A549 and HCT116 cells received treatment of cisplatin in different concentrations with or without Fer-1 (0.5 μmol/L) as displayed. Cell viabilities were analyzed by MTT. Standard error represents three independent experiments (n=3). *p < 0.05, **p < 0.01, Student’s t test.
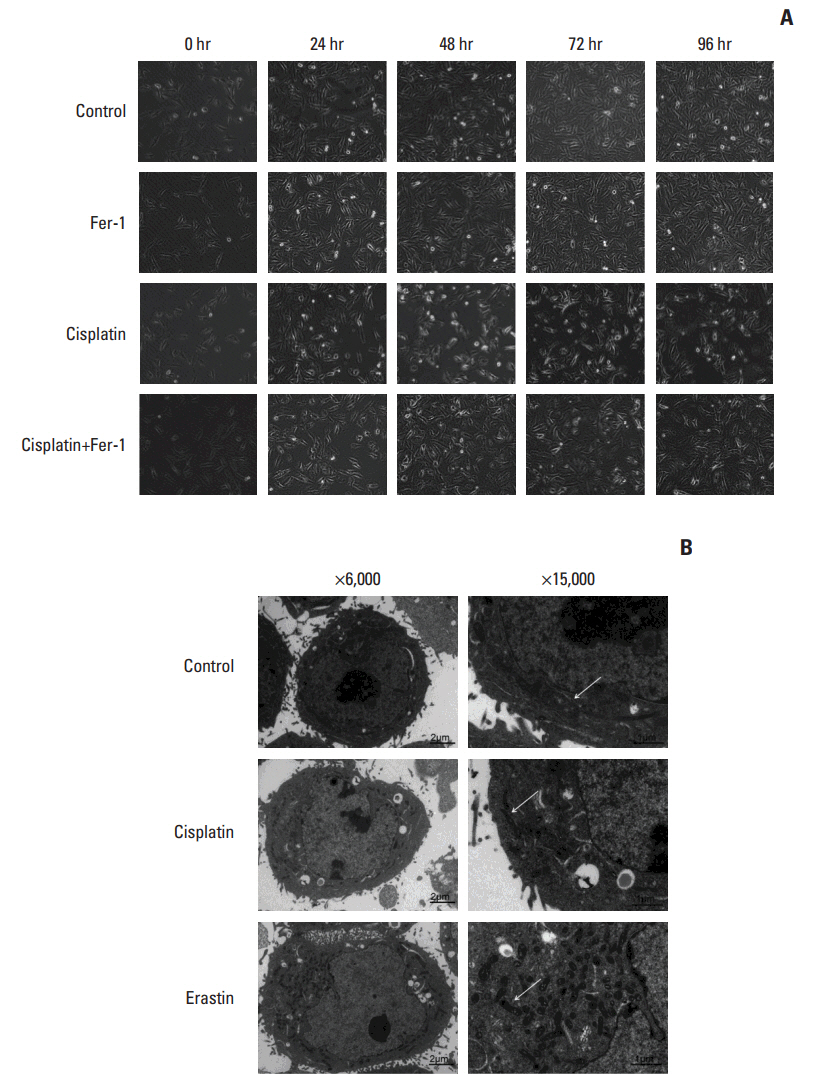
Fig. 2. Microscopy images of cisplatin treated cells. (A) Optical microscopy images of A549 cells under the treatment of cisplatin (5 μg/mL) with or without ferrostatin-1 (Fer-1, 0.5 μmol/L) for indicated time (×100, scale bars=100 μm). (B) Transmission electron microscopy images for HCT116 cells which were treated with cisplatin (5 μg/mL) and erastin (10 μmol/L), respectively for 48 hours. Arrows indicate mitochondria in cells (left: ×6,000, scale bars=2 μm; right: ×15,000, scale bars=1 μm).
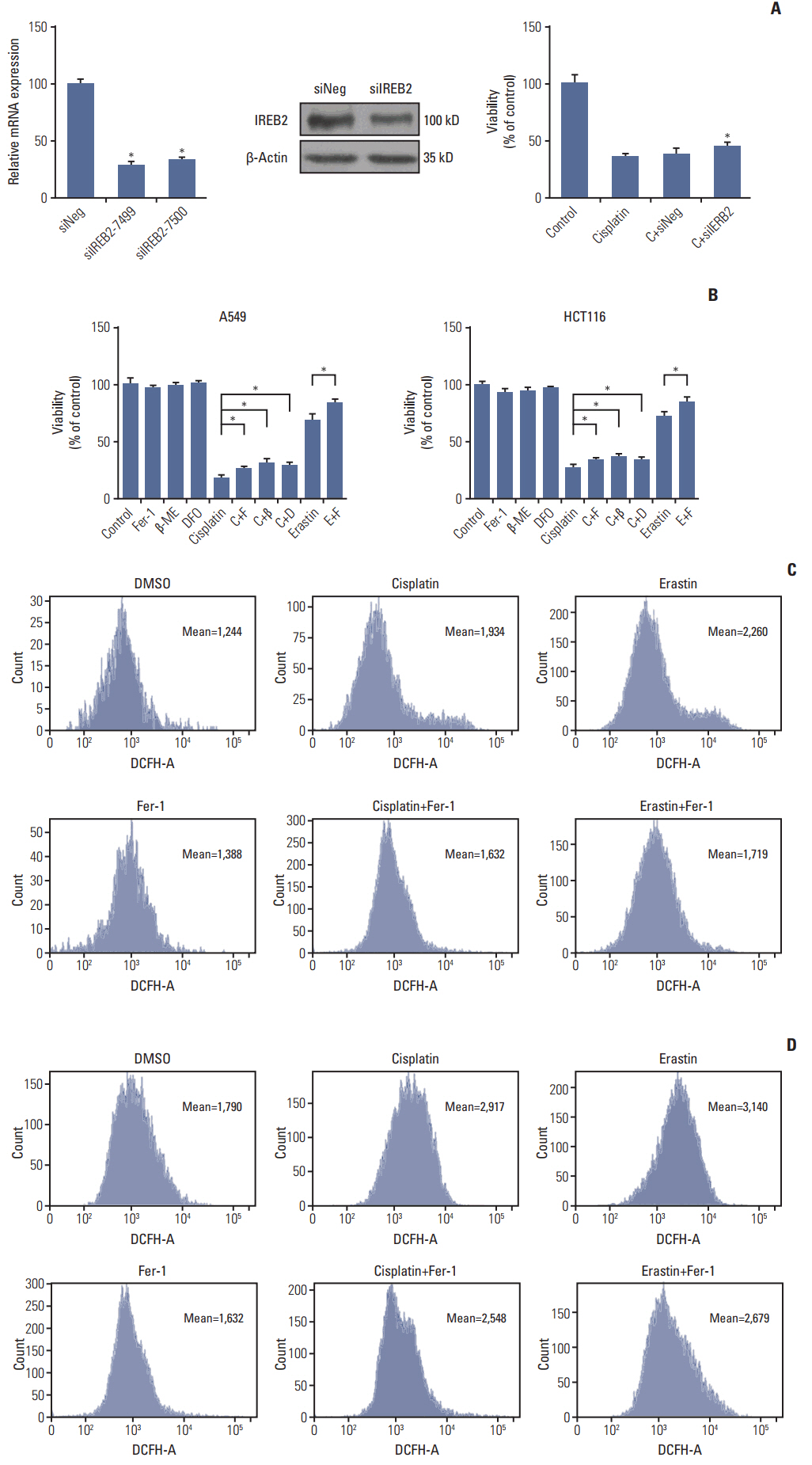
Fig. 3. Cisplatin induced ferroptosis in A549 and HCT116 cells. (A) Polymerase chain reaction and western blot analysis of HCT116 cells transferred with small interfering RNAs (siRNAs) targeting IREB2 (siIERB2). HCT116 cells were transferred with siIERB2 or siNeg 24 hours in advance, then, treated with cisplatin (5 μg/mL) or normal saline for 48 hours. Cell viabilities were analyzed by MTT. (B) A549 and HCT116 cells were under the treatment of cisplatin (5 μg/mL) for 48 hours with different specific ferroptosis inhibitors. Cell viabilities were analyzed by MTT. Standard error represents three independent experiments (n=3). *p < 0.05, Student’s t test. C, cisplatin; F, ferrostatin-1 (Fer-1, 0.5 μmol/L); β, β-mercaptoethanol (β-ME,=50 μmol/L); D, deferoxamine (DFO, 50 μmol/L); E, erastin (10 μmol/L). (C) A549 cells were under the treatment of cisplatin (5 μg/mL), erastin (10 μmol/L), or Fer-1 (0.5 μmol/L) as demonstrated for 48 hours. Reactive oxygen species (ROS) levels in cells were evaluated and exhibited. (D) HCT116 cells were under the treatment of cisplatin (5 μg/mL), erastin (10 μmol/L), or Fer-1 (0.5 μmol/L) as demonstrated for 48 hours. ROS levels in cells were evaluated and exhibited.
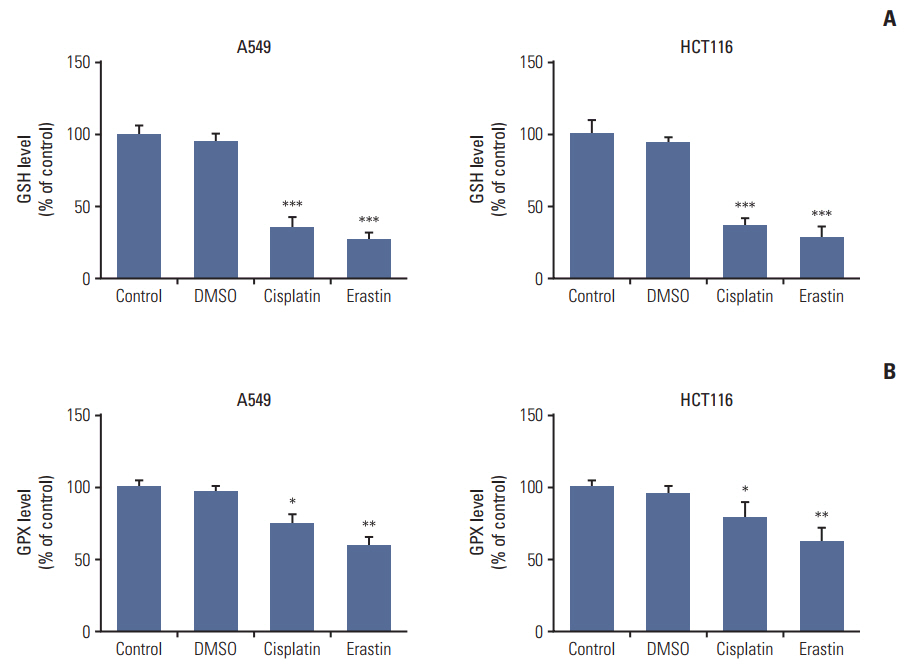
Fig. 4. Cisplatin led to intracellular glutathione (GSH) depletion and glutathione peroxidases (GPXs) inactivation in A549 and HCT116 cells. (A) GSH level analysis of A549 and HCT116 cells. (B) GPXs activity analysis of A549 and HCT116 cells. Cells were treated with cisplatin (5 μg/mL) or erastin (10 μmol/L, as a positive control) for 48 hours, respectively. DMSO, dimethyl sulfoxide. Standard error represents three independent experiments (n=3). *p < 0.05, **p < 0.01, ***p < 0.001, Student’s t test.

Fig. 5. Improved anti-tumor activity was observed in combination of cisplatin and erastin. (A) A549 cells were treated with cisplatin and erastin of different concentrations as demonstrated for 48 hours. (B) HCT116 cells were treated with cisplatin and erastin of different concentrations as demonstrated for 48 hours. (C) A549 and HCT116 cells were under the treatment of cisplatin (5 μg/mL) or erastin (10 μmol/L) for 48 hours, together with z-vad-fmk (20 μmol/L) or ferrostatin-1 (Fer-1, 0.5 μmol/L), respectively. Cell viabilities were analyzed by MTT. C, cisplatin; E, erastin; F, ferrostatin-1 (Fer-1); Z, z-vad-fmk; β-ME, β-mercaptoethanol; n.s., not significant. Standard error represents three independent experiments (n=3). **p < 0.01, ***p < 0.001, Student’s t test. (D) Optical microscopy images of A549 cells under the treatment of cisplatin (5 μg/mL), erastin (10 μmol/L) with or without β-ME (50 μmol/L) for indicated time (×100, scale bars=100 μm). (E) HCT116 cells were under the treatment of cisplatin (5 μg/mL), erastin (10 μmol/L) with or without β-ME (50 μmol/L) as demonstrated for 48 hours. Reactive oxygen species levels in cells were evaluated and exhibited.
Reference
-
References
1. Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009; 16:3–11.
Article2. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. 2003; 3:285–96.
Article3. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009; 7:99–109.
Article4. Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol. 2010; 22:263–8.
Article5. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012; 149:1060–72.
Article6. Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007; 447:864–8.
Article7. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008; 15:234–45.
Article8. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014; 10:9–17.
Article9. Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001; 15:1633–40.10. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014; 156:317–31.
Article11. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016; 23:369–79.
Article12. Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer. 2013; 133:1732–42.
Article13. Louandre C, Marcq I, Bouhlal H, Lachaier E, Godin C, Saidak Z, et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015; 356(2 Pt B):971–7.
Article14. Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science. 2009; 326:722–6.
Article15. Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science. 2009; 326:718–21.
Article16. Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016; 73:2195–209.
Article17. Wang X, Guo Z. The role of sulfur in platinum anticancer chemotherapy. Anticancer Agents Med Chem. 2007; 7:19–34.
Article18. Min Y, Mao CQ, Chen S, Ma G, Wang J, Liu Y. Combating the drug resistance of cispla-tin using a platinum prodrug based delivery system. Angew Chem Int Ed Engl. 2012; 51:6742–7.
Article19. Wang D, Lippard SJ. Cellular processing of platinum anti-cancer drugs. Nat Rev Drug Discov. 2005; 4:307–20.
Article20. Zamble DB, Lippard SJ. Cisplatin and DNA repair in cancer chemotherapy. Trends Biochem Sci. 1995; 20:435–9.
Article21. Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res. 2016; 106:27–36.
Article22. Richon VM, Schulte N, Eastman A. Multiple mechanisms of resistance to cis-diamminedichloroplatinum(II) in murine leukemia L1210 cells. Cancer Res. 1987; 47:2056–61.23. Ferry KV, Hamilton TC, Johnson SW. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: role of ERCC1-XPF. Biochem Pharmacol. 2000; 60:1305–13.24. Roh JL, Kim EH, Jang HJ, Park JY, Shin D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016; 381:96–103.
Article25. Mascaux C, Iannino N, Martin B, Paesmans M, Berghmans T, Dusart M, et al. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br J Cancer. 2005; 92:131–9.
Article26. Cengel KA, Voong KR, Chandrasekaran S, Maggiorella L, Brunner TB, Stanbridge E, et al. Oncogenic K-Ras signals through epidermal growth factor receptor and wild-type H-Ras to promote radiation survival in pancreatic and colorectal carcinoma cells. Neoplasia. 2007; 9:341–8.
Article27. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005; 2:e17.
Article28. Massarelli E, Varella-Garcia M, Tang X, Xavier AC, Ozburn NC, Liu DD, et al. KRAS mutation is an important predictor of resistance to therapy with epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. Clin Cancer Res. 2007; 13:2890–6.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Ferroptosis Is Crucial for Cisplatin Induced Sertoli Cell Injury via N6-Methyladenosine Dependent Manner
- Temozolomide Drives Ferroptosis via a DMT1-Dependent Pathway in Glioblastoma Cells
- Immunotherapy with methyl gallate, an inhibitor of Treg cell migration, enhances the anti-cancer effect of cisplatin therapy
- Protaetia brevitarsis larvae extract protects against lipopolysaccharidesinduced ferroptosis and inflammation by inhibiting acid sphingomyelinase
- Ferroptosis and its role in gastric and colorectal cancers
