J Clin Neurol.
2018 Jan;14(1):22-28. 10.3988/jcn.2018.14.1.22.
Efficacy of Stiripentol in Dravet Syndrome with or without SCN1A Mutations
- Affiliations
-
- 1Divison of Pediatric Neurology, Department of Pediatrics, Severance Children's Hospital, Yonsei University College of Medicine, Epilepsy Research Institute, Seoul, Korea. hipo0207@yuhs.ac
- 2Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Korea.
- 3Department of Pediatrics, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea.
- 4Department of Pediatrics, National Health Insurance Service Ilsan Hospital, Goyang, Korea.
- 5Department of Biotechnology, Korea University, Seoul, Korea.
- KMID: 2399595
- DOI: http://doi.org/10.3988/jcn.2018.14.1.22
Abstract
- BACKGROUND AND PURPOSE
The aim of this study was to determine the effectiveness of stiripentol (STP) add-on therapy to valproate and clobazam in patients with Dravet syndrome (DS) according to the presence of mutations in the sodium channel alpha-1 subunit gene (SCN1A).
METHODS
We performed direct sequencing to analyze SCN1A mutations in 32 patients with clinically confirmed with DS, and classified them into mutation (pathogenic or likely pathogenic) and nonmutation groups based on American College of Medical Genetics and Genomics guidelines. We compared the efficacy of STP in reducing the seizure frequency between the two groups.
RESULTS
The 32 patients comprised 15 patients in the mutation group (with definite SCN1A mutations) and 17 patients in the nonmutation group with variants of unknown significance or benign variants. The clinical profile did not differ significantly between the mutation and nonmutation groups. The seizure frequency relative to baseline reduced by 72.53±23.00% (mean±SD) in the mutation group versus 50.58±40.14% in the nonmutation group (p=0.004). The efficacy of STP was better in DS patients with missense mutations that in those with truncation mutations, and was not favorable in patients with mutations at linkers between domains (DII-DIII), linkers between segments of domain I (DI S1-S2), or splice sites, although the small number of patients prevented statistical analyses.
CONCLUSIONS
The efficacy of STP was significantly better in DS patients with definite SCN1A mutations than in those without mutations.
MeSH Terms
Figure
-
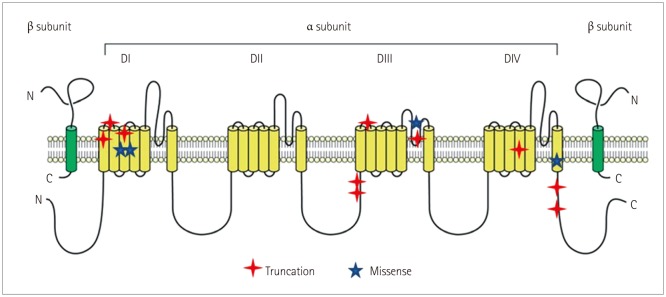
Fig. 1 Protein locations of sodium channel alpha-1 subunit gene (SCN1A) mutations.
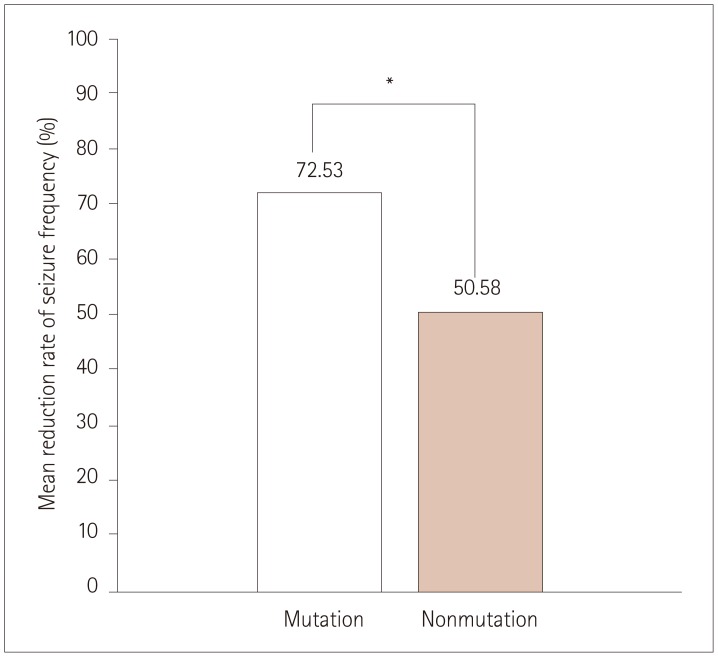
Fig. 2 Efficacy of stiripentol according to the presence of SCN1A mutations. *p=0.004. SCN1A: sodium channel alpha-1 subunit gene.
Reference
-
1. Connolly MB. Dravet syndrome: diagnosis and long-term course. Can J Neurol Sci. 2016; 43(Suppl 3):S3–S8.
Article2. Dravet C, Bureau M, Oguni H, Fukuyama Y, Cokar O. Severe myoclonic epilepsy in infants (Dravet syndrome). In : Roger J, Bureau M, Dravet C, Genton P, Tassinari C, Wolf P, editors. Epileptic Syndromes in Infancy. Childhood and Adolescence. 3rd ed. London: John Libbey;2002. p. 81–103.3. Ceulemans BP, Claes LR, Lagae LG. Clinical correlations of mutations in the SCN1A gene: from febrile seizures to severe myoclonic epilepsy in infancy. Pediatr Neurol. 2004; 30:236–243. PMID: 15087100.
Article4. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001; 68:1327–1332. PMID: 11359211.
Article5. Wallace RH, Hodgson BL, Grinton BE, Gardiner RM, Robinson R, Rodriguez-Casero V, et al. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003; 61:765–769. PMID: 14504318.6. Kearney J. The more, the better: modeling dravet syndrome with induced pluripotent stem cell-derived neurons. Epilepsy Curr. 2014; 14:33–34. PMID: 24526875.
Article7. Aras LM, Isla J, Mingorance-Le Meur A. The European patient with Dravet syndrome: results from a parent-reported survey on antiepileptic drug use in the European population with Dravet syndrome. Epilepsy Behav. 2015; 44:104–109. PMID: 25666511.
Article8. Quilichini PP, Chiron C, Ben-Ari Y, Gozlan H. Stiripentol, a putative antiepileptic drug, enhances the duration of opening of GABA-A receptor channels. Epilepsia. 2006; 47:704–716. PMID: 16650136.9. Fisher JL. The effects of stiripentol on GABA(A) receptors. Epilepsia. 2011; 52(Suppl 2):76–78. PMID: 21463286.
Article10. Brigo F, Storti M. Antiepileptic drugs for the treatment of severe myoclonic epilepsy in infancy. Cochrane Database Syst Rev. 2013; CD010483. PMID: 24254932.
Article11. Marini C, Scheffer IE, Nabbout R, Suls A, De Jonghe P, Zara F, et al. The genetics of Dravet syndrome. Epilepsia. 2011; 52(Suppl 2):24–29. PMID: 21463275.
Article12. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. PMID: 25741868.
Article13. Takayama R, Fujiwara T, Shigematsu H, Imai K, Takahashi Y, Yamakawa K, et al. Long-term course of Dravet syndrome: a study from an epilepsy center in Japan. Epilepsia. 2014; 55:528–538. PMID: 24502503.
Article14. Balestrini S, Sisodiya SM. Audit of use of stiripentol in adults with Dravet syndrome. Acta Neurol Scand. 2017; 135:73–79. PMID: 27231140.
Article15. Inoue Y, Ohtsuka Y;. Long-term safety and efficacy of stiripentol for the treatment of Dravet syndrome: A multicenter, open-label study in Japan. Epilepsy Res. 2015; 113:90–97. PMID: 25986195.
Article16. Inoue Y, Ohtsuka Y;. Effectiveness of add-on stiripentol to clobazam and valproate in Japanese patients with Dravet syndrome: additional supportive evidence. Epilepsy Res. 2014; 108:725–731. PMID: 24630050.
Article17. Inoue Y, Ohtsuka Y, Oguni H, Tohyama J, Baba H, Fukushima K, et al. Stiripentol open study in Japanese patients with Dravet syndrome. Epilepsia. 2009; 50:2362–2368. PMID: 19552653.
Article18. Chiron C, Marchand MC, Tran A, Rey E, d'Athis P, Vincent J, et al. Stiripentol in severe myoclonic epilepsy in infancy: a randomised placebo-controlled syndrome-dedicated trial. STICLO study group. Lancet. 2000; 356:1638–1642. PMID: 11089822.19. Wirrell EC, Laux L, Franz DN, Sullivan J, Saneto RP, Morse RP, et al. Stiripentol in Dravet syndrome: results of a retrospective U.S. study. Epilepsia. 2013; 54:1595–1604. PMID: 23848835.
Article20. Perez J, Chiron C, Musial C, Rey E, Blehaut H, d'Athis P, et al. Stiripentol: efficacy and tolerability in children with epilepsy. Epilepsia. 1999; 40:1618–1626. PMID: 10565591.
Article21. Strzelczyk A, Schubert-Bast S, Reese JP, Rosenow F, Stephani U, Boor R. Evaluation of health-care utilization in patients with Dravet syndrome and on adjunctive treatment with stiripentol and clobazam. Epilepsy Behav. 2014; 34:86–91. PMID: 24727467.
Article22. De Liso P, Chemaly N, Laschet J, Barnerias C, Hully M, Leunen D, et al. Patients with Dravet syndrome in the era of stiripentol: a French cohort cross-sectional study. Epilepsy Res. 2016; 125:42–46. PMID: 27389706.
Article23. Ishii A, Watkins JC, Chen D, Hirose S, Hammer MF. Clinical implications of SCN1A missense and truncation variants in a large Japanese cohort with Dravet syndrome. Epilepsia. 2017; 58:282–290. PMID: 28012175.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- SCN1A Variants in Patients with Dravet Syndrome
- The Genetic Facets of Dravet Syndrome: Recent Insights
- SCN1A Gene Mutation and Adaptive Functioning in 18 Vietnamese Children with Dravet Syndrome
- Mutations of SCN1A in Familial Febrile Seizures
- Current Pharmacologic Strategies for Treatment of Intractable Epilepsy in Children
