J Clin Neurol.
2017 Jan;13(1):62-70. 10.3988/jcn.2017.13.1.62.
SCN1A Gene Mutation and Adaptive Functioning in 18 Vietnamese Children with Dravet Syndrome
- Affiliations
-
- 1Research Center for Genetics and Reproductive Health, School of Medicine, Vietnam National University HCMC, Ho Chi Minh City, Vietnam. hangdo009@gmail.com
- 2Center for Molecular Biomedicine, University of Medicine and Pharmacy HCMC, Ho Chi Minh City, Vietnam.
- 3Neurology Department, Children Hospital 2, Ho Chi Minh City, Vietnam.
- 4Department of Herbal Medicine Resource, Institute of Bioscience and Biotechnology, Kangwon National University, Samcheok, Korea.
- 5Department of Laboratory Medicine, Faculty of Nursing and Medical Technology, Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
- 6National Institute of Forensic Medicine, Hanoi, Vietnam.
- KMID: 2364899
- DOI: http://doi.org/10.3988/jcn.2017.13.1.62
Abstract
- BACKGROUND AND PURPOSE
Dravet syndrome is a rare and severe type of epilepsy in infants. The heterogeneity in the overall intellectual disability that these patients suffer from has been attributed to differences in genetic background and epilepsy severity.
METHODS
Eighteen Vietnamese children diagnosed with Dravet syndrome were included in this study. SCN1A variants were screened by direct sequencing and multiplex ligation-dependent probe amplification. Adaptive functioning was assessed in all patients using the Vietnamese version of the Vineland Adaptive Behavior Scales, and the results were analyzed relative to the SCN1A variants and epilepsy severity.
RESULTS
We identified 13 pathogenic or likely pathogenic variants, including 6 that have not been reported previously. We found no correlations between the presence or type of SCN1A variants and the level of adaptive functioning impairment or severity of epilepsy. Only two of nine patients aged at least 5 years had an adaptive functioning score higher than 50. Both of these patients had a low frequency of convulsive seizures and no history of status epilepticus or prolonged seizures. The remaining seven had very low adaptive functioning scores (39 or less) despite the variability in the severity of their epilepsy confirming the involvement of factors other than the severity of epilepsy in determining the developmental outcome.
CONCLUSIONS
Our study expands the spectrum of known SCN1A variants and confirms the current understanding of the role of the genetic background and epilepsy severity in determining the developmental outcome of Dravet syndrome patients.
Keyword
MeSH Terms
Figure
-
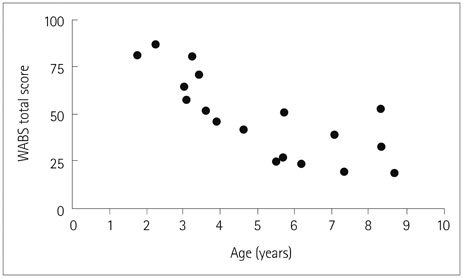
Fig. 1 Adaptive functioning as assessed using the Vietnamese version of the Vineland Adaptive Behavior Scales (VVABS) in all 18 included patients during their last visit.
Reference
-
1. Dravet C. The core Dravet syndrome phenotype. Epilepsia. 2011; 52:Suppl 2. 3–9.
Article2. Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001; 68:1327–1332.
Article3. Scheffer IE. Diagnosis and long-term course of Dravet syndrome. Eur J Paediatr Neurol. 2012; 16:Suppl 1. S5–S8.
Article4. Carvill GL, Weckhuysen S, McMahon JM, Hartmann C, Møller RS, Hjalgrim H, et al. GABRA1 and STXBP1: novel genetic causes of Dravet syndrome. Neurology. 2014; 82:1245–1253.
Article5. Suls A, Jaehn JA, Kecskés A, Weber Y, Weckhuysen S, Craiu DC, et al. De novo loss-of-function mutations in CHD2 cause a fever-sensitive myoclonic epileptic encephalopathy sharing features with Dravet syndrome. Am J Hum Genet. 2013; 93:967–975.
Article6. Parihar R, Ganesh S. The SCN1A gene variants and epileptic encephalopathies. J Hum Genet. 2013; 58:573–580.
Article7. Lim BC, Hwang H, Chae JH, Choi JE, Hwang YS, Kang SH, et al. SCN1A mutational analysis in Korean patients with Dravet syndrome. Seizure. 2011; 20:789–794.
Article8. Kwong AK, Fung CW, Chan SY, Wong VC. Identification of SCN1A and PCDH19 mutations in Chinese children with Dravet syndrome. PLoS One. 2012; 7:e41802.
Article9. Chieffo D, Battaglia D, Lettori D, Del Re M, Brogna C, Dravet C, et al. Neuropsychological development in children with Dravet syndrome. Epilepsy Res. 2011; 95:86–93.
Article10. Brunklaus A, Ellis R, Reavey E, Forbes GH, Zuberi SM, et al. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain. 2012; 135(Pt 8):2329–2336.
Article11. Ragona F, Granata T, Dalla Bernardina B, Offredi F, Darra F, Battaglia D, et al. Cognitive development in Dravet syndrome: a retrospective, multicenter study of 26 patients. Epilepsia. 2011; 52:386–392.
Article12. Brunklaus A, Zuberi SM. Dravet syndrome--from epileptic encephalopathy to channelopathy. Epilepsia. 2014; 55:979–984.
Article13. Guerrini R, Oguni H. Borderline Dravet syndrome: a useful diagnostic category? Epilepsia. 2011; 52:Suppl 2. 10–12.
Article14. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424.
Article15. Goldberg MR, Dill CA, Shin JY, Nguyen VN. Reliability and validity of the Vietnamese Vineland Adaptive Behavior Scales with preschool-age children. Res Dev Disabil. 2009; 30:592–602.
Article16. Depienne C, Trouillard O, Saint-Martin C, Gourfinkel-An I, Bouteiller D, Carpentier W, et al. Spectrum of SCN1A gene mutations associated with Dravet syndrome: analysis of 333 patients. J Med Genet. 2009; 46:183–191.
Article17. Oguni H, Hayashi K, Awaya Y, Fukuyama Y, Osawa M. Severe myoclonic epilepsy in infants--a review based on the Tokyo Women's Medical University series of 84 cases. Brain Dev. 2001; 23:736–748.
Article18. Kobayashi K, Ohmori I, Ouchida M, Ohtsuka Y. Dravet syndrome with an exceptionally good seizure outcome in two adolescents. Epileptic Disord. 2011; 13:340–344.
Article19. Akiyama M, Kobayashi K, Yoshinaga H, Ohtsuka Y. A long-term follow-up study of Dravet syndrome up to adulthood. Epilepsia. 2010; 51:1043–1052.
Article20. Kanai K, Hirose S, Oguni H, Fukuma G, Shirasaka Y, Miyajima T, et al. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology. 2004; 63:329–334.
Article21. Zuberi SM, Brunklaus A, Birch R, Reavey E, Duncan J, Forbes GH. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology. 2011; 76:594–600.
Article22. Marini C, Mei D, Temudo T, Ferrari AR, Buti D, Dravet C, et al. Idiopathic epilepsies with seizures precipitated by fever and SCN1A abnormalities. Epilepsia. 2007; 48:1678–1685.
Article23. Wolff M, Cassé-Perrot C, Dravet C. Severe myoclonic epilepsy of infants (Dravet syndrome): natural history and neuropsychological findings. Epilepsia. 2006; 47:Suppl 2. 45–48.
Article24. Ragona F, Brazzo D, De Giorgi I, Morbi M, Freri E, Teutonico F, et al. Dravet syndrome: early clinical manifestations and cognitive outcome in 37 Italian patients. Brain Dev. 2010; 32:71–77.
Article25. Villeneuve N, Laguitton V, Viellard M, Lépine A, Chabrol B, Dravet C, et al. Cognitive and adaptive evaluation of 21 consecutive patients with Dravet syndrome. Epilepsy Behav. 2014; 31:143–148.
Article26. Yu MJ, Shi YW, Gao MM, Deng WY, Liu XR, Chen L, et al. Milder phenotype with SCN1A truncation mutation other than SMEI. Seizure. 2010; 19:443–445.
Article27. Gennaro E, Veggiotti P, Malacarne M, Madia F, Cecconi M, Cardinali S, et al. Familial severe myoclonic epilepsy of infancy: truncation of Nav1.1 and genetic heterogeneity. Epileptic Disord. 2003; 5:21–25.28. Dravet C. How Dravet syndrome became a model for studying childhood genetic epilepsies. Brain. 2012; 135(Pt 8):2309–2311.
Article29. Catarino CB, Liu JY, Liagkouras I, Gibbons VS, Labrum RW, Ellis R, et al. Dravet syndrome as epileptic encephalopathy: evidence from long-term course and neuropathology. Brain. 2011; 134(Pt 10):2982–3010.
Article30. Bender AC, Natola H, Ndong C, Holmes GL, Scott RC, Lenck-Santini PP. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis. 2013; 54:297–307.
Article31. Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013; 8:176.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- SCN1A Variants in Patients with Dravet Syndrome
- Efficacy of Stiripentol in Dravet Syndrome with or without SCN1A Mutations
- Effects of Cannabidiol on Adaptive Behavior and Quality of Life in Pediatric Patients With Treatment-Resistant Epilepsy
- Mutations of SCN1A in Familial Febrile Seizures
- The Genetic Facets of Dravet Syndrome: Recent Insights
