Impaired Nucleoporins Are Present in Sporadic Amyotrophic Lateral Sclerosis Motor Neurons that Exhibit Mislocalization of the 43-kDa TAR DNA-Binding Protein
- Affiliations
-
- 1Department of Neurology, Tokyo Medical University, Tokyo, Japan. haizawa@tokyo-med.ac.jp
- 2Division of Clinical Biotechnology, Center for Disease Biology and Integrative Medicine, Graduate School of Medicine, The University of Tokyo, Tokyo, Japan.
- 3Department of Neurology, Asahikawa Medical Center, National Hospital Organization, Asahikawa, Japan.
- KMID: 2451146
- DOI: http://doi.org/10.3988/jcn.2019.15.1.62
Abstract
- BACKGROUND AND PURPOSE
Disruption of nucleoporins has been reported in the motor neurons of patients with sporadic amyotrophic lateral sclerosis (sALS). However, the precise changes in the morphology of nucleoporins associated with the pathology of the 43-kDa TAR DNA-binding protein (TDP-43) in the disease process remain unknown. We investigated the expression of nucleoporins that constitute the nuclear pore complex (NPC) in spinal motor neurons that exhibit sALS in relation to TDP-43 pathology, which is a reliable neuropathological hallmark of sALS.
METHODS
Paraffin-embedded sections of the lumbar spinal cord were obtained for immunofluorescence analysis from seven control subjects and six sALS patients. Anti-TDP-43 antibody, anti-nucleoporin p62 (NUP62) antibody, and anti-karyopherin beta 1 (KPNB1) antibody were applied as primary antibodies, and then visualized using appropriate secondary antibodies. The sections were then examined under a fluorescence microscope.
RESULTS
NUP62 and KPNB1 immunoreactivity appeared as a smooth round rim bordering the nuclear margin in normal spinal motor neurons that exhibited nuclear TDP-43 immunoreactivity. sALS spinal motor neurons with apparent TDP-43 mislocalization demonstrated irregular, disrupted nuclear staining for NUP62 or KPNB1. Some atrophic sALS spinal motor neurons with TDP-43 mislocalization presented no NUP62 immunoreactivity.
CONCLUSIONS
Our findings suggest a close relationship between NPC alterations and TDP-43 pathology in the degenerative process of the motor neurons of sALS patients.
Keyword
MeSH Terms
Figure
-
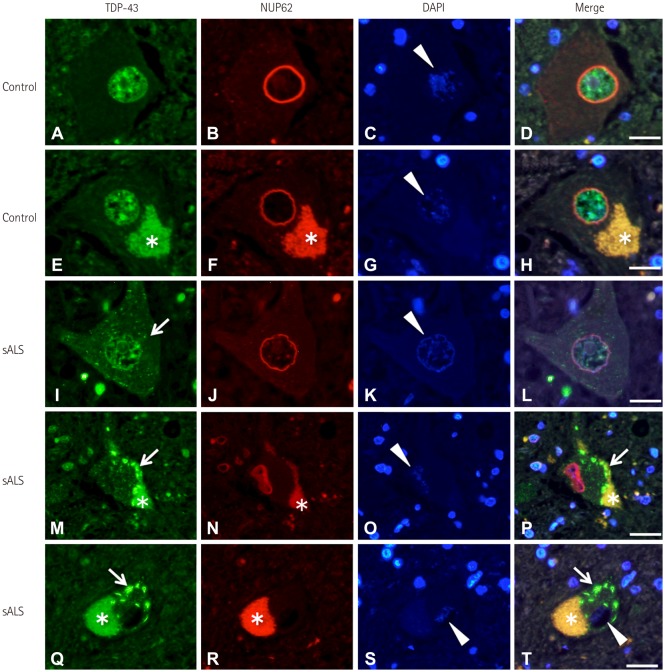
Fig. 1 TDP-43 and NUP62 immunoreactivity in control and sALS spinal motor neurons. TDP-43 was localized to the nucleus (arrowheads) and NUP62 immunoreactivity was present along the nuclear membrane in control spinal motor neurons (A–H). Motor neurons with decreased nuclear TDP-43 (arrowhead) exhibited fine TDP-43-positive dots (arrow) within the cytoplasm in sALS (I–L). NUP62 was present in the disrupted nuclear membrane of sALS spinal motor neurons with apparent TDP-43 mislocalization (arrows, arrowhead) (M–P). NUP62 immunoreactivity was absent (arrowheads) in some of the atrophic sALS spinal motor neurons with apparent TDP-43 mislocalization (arrows) (Q–T). Scale bars indicate 30 m. *Lipofuscin. DAPI: 4′,6-diamidino-2-phenylindole, NUP62: nucleoporin p62, sALS: sporadic amyotrophic lateral sclerosis, TDP-43: 43-kDa TAR DNA-binding protein.
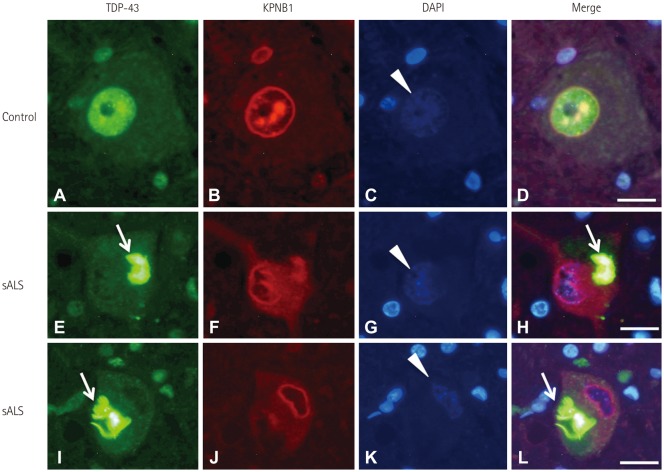
Fig. 2 TDP-43 and KPNB1 localization in control and ALS spinal motor neurons. TDP-43 was localized in the nucleus (arrowhead), and KPNB1 labeling was present along the nuclear membrane and appeared as dots in the nucleus of control spinal motor neurons (A–D). KPNB1 labeling was present in the disrupted or deformed nuclear membrane of sALS spinal motor neurons with TDP-43 mislocalization (arrows, arrowheads) (E–L). Scale bars indicate 30 m. DAPI: 4′,6-diamidino-2-phenylindole, KPNB1: karyopherin beta 1, sALS: sporadic amyotrophic lateral sclerosis, TDP-43: 43-kDa TAR DNA-binding protein.
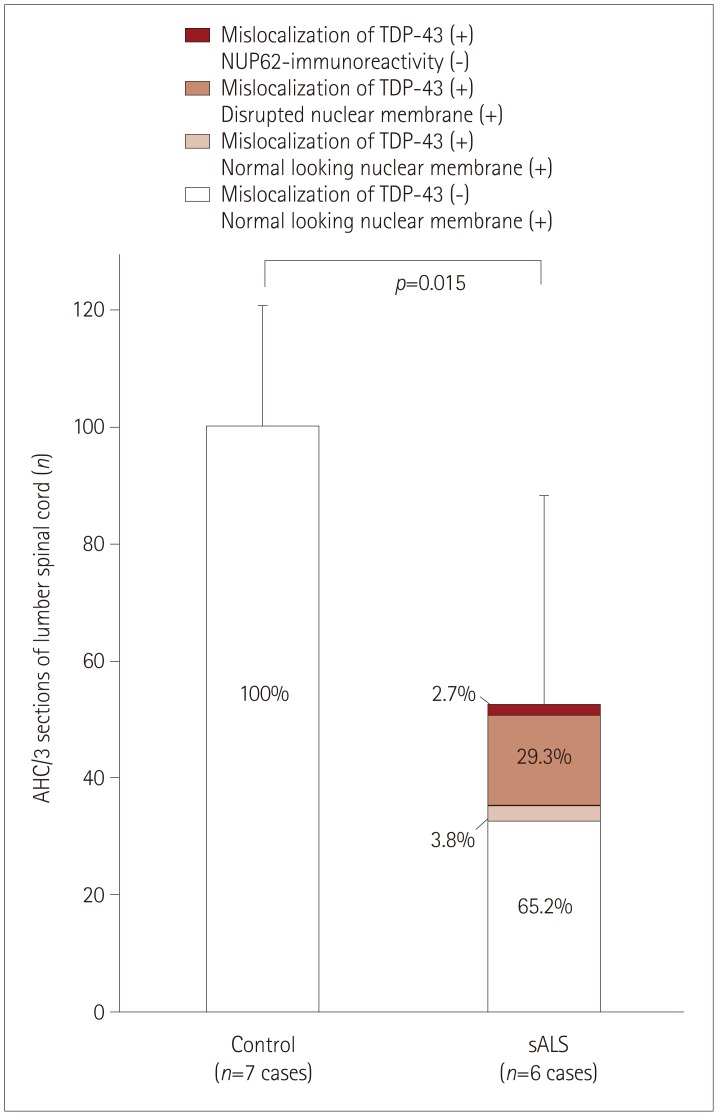
Fig. 3 Quantification of spinal motor neurons in lumbar spinal-cord tissue from control and sALS cases using double immunofluorescence labeling of NUP62 antibodies and TDP-43 antibodies. There were fewer AHC in sALS patients (52.2±36.4) than in control subjects (104.6±15.8) (p=0.015). Spinal motor neurons in control spinal anterior horns exhibited nuclear membranes that were labeled with anti-NUP62 antibodies and did not show TDP-43 mislocalization. In sALS spinal anterior horns, approximately two-thirds of the motor neurons exhibited normal round membranes without TDP-43-positive cytoplasmic inclusions. The remaining 34.8% of neurons in sALS samples exhibited TDP-43 mislocalization: 3.8% exhibited normal nuclear NUP62 immunoreactivity, 29.3% demonstrated disrupted NUP62 nuclear staining, and 2.7% were negative for NUP62 immunoreactivity. AHC: anterior horn cells, NUP62: nucleoporin p62, sALS: sporadic amyotrophic lateral sclerosis, TDP-43: 43-kDa TAR DNA-binding protein.
Reference
-
1. Kim HJ, Taylor JP. Lost in transportation: nucleocytoplasmic transport defects in ALS and other neurodegenerative diseases. Neuron. 2017; 96:285–297. PMID: 29024655.
Article2. Jovičić A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat Neurosci. 2015; 18:1226–1229. PMID: 26308983.
Article3. Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015; 525:129–133. PMID: 26308899.
Article4. Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015; 525:56–61. PMID: 26308891.
Article5. Kinoshita Y, Ito H, Hirano A, Fujita K, Wate R, Nakamura M, et al. Nuclear contour irregularity and abnormal transporter protein distribution in anterior horn cells in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2009; 68:1184–1192. PMID: 19816199.
Article6. Nagara Y, Tateishi T, Yamasaki R, Hayashi S, Kawamura M, Kikuchi H, et al. Impaired cytoplasmic-nuclear transport of hypoxia-inducible factor-1α in amyotrophic lateral sclerosis. Brain Pathol. 2013; 23:534–546. PMID: 23368766.
Article7. Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018; 21:228–239. PMID: 29311743.
Article8. Kaneb HM, Folkmann AW, Belzil VV, Jao LE, Leblond CS, Girard SL, et al. Deleterious mutations in the essential mRNA metabolism factor, hGle1, in amyotrophic lateral sclerosis. Hum Mol Genet. 2015; 24:1363–1373. PMID: 25343993.
Article9. Hideyama T, Yamashita T, Suzuki T, Tsuji S, Higuchi M, Seeburg PH, et al. Induced loss of ADAR2 engenders slow death of motor neurons from Q/R site-unedited GluR2. J Neurosci. 2010; 30:11917–11925. PMID: 20826656.
Article10. Yamashita T, Aizawa H, Teramoto S, Akamatsu M, Kwak S. Calpain-dependent disruption of nucleo-cytoplasmic transport in ALS motor neurons. Sci Rep. 2017; 7:39994. PMID: 28045133.
Article11. Takuma H, Kwak S, Yoshizawa T, Kanazawa I. Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann Neurol. 1999; 46:806–815. PMID: 10589532.
Article12. Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004; 427:801. PMID: 14985749.13. Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, et al. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012; 45:1121–1128. PMID: 22226999.
Article14. Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006; 351:602–611. PMID: 17084815.
Article15. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006; 314:130–133. PMID: 17023659.
Article16. Aizawa H, Sawada J, Hideyama T, Yamashita T, Katayama T, Hasebe N, et al. TDP-43 pathology in sporadic ALS occurs in motor neurons lacking the RNA editing enzyme ADAR2. Acta Neuropathol. 2010; 120:75–84. PMID: 20372915.
Article17. Yamashita T, Hideyama T, Hachiga K, Teramoto S, Takano J, Iwata N, et al. A role for calpain-dependent cleavage of TDP-43 in amyotrophic lateral sclerosis pathology. Nat Commun. 2012; 3:1307. PMID: 23250437.
Article18. Lott K, Cingolani G. The importin β binding domain as a master regulator of nucleocytoplasmic transport. Biochim Biophys Acta. 2011; 1813:1578–1592. PMID: 21029753.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- The Pathogenetic Role of TAR DNA Binding Protein (TDP-43) in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia
- Prolyl Isomerase Pin1 Expression in the Spinal Motor Neurons of Patients With Sporadic Amyotrophic Lateral Sclerosis
- An Autopsy Confirmed Case of Amyotrophic Lateral Sclerosis with TDP Pathology
- Increased Neuronal and Glial Poly (ADP-Ribose) Polymerase Immunoreactivity in the Brain of Sporadic Amyotrophic Lateral Sclerosis
- Diagnosis and management of amyotrophic lateral sclerosis
