Genetic regulation of linear growth
- Affiliations
-
- 1Pediatric Endocrine, Metabolism and Genetics, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA. jeeyh@mail.nih.gov
- KMID: 2447370
- DOI: http://doi.org/10.6065/apem.2019.24.1.2
Abstract
- Linear growth occurs at the growth plate. Therefore, genetic defects that interfere with the normal function of the growth plate can cause linear growth disorders. Many genetic causes of growth disorders have already been identified in humans. However, recent genome-wide approaches have broadened our knowledge of the mechanisms of linear growth, not only providing novel monogenic causes of growth disorders but also revealing single nucleotide polymorphisms in genes that affect height in the general population. The genes identified as causative of linear growth disorders are heterogeneous, playing a role in various growth-regulating mechanisms including those involving the extracellular matrix, intracellular signaling, paracrine signaling, endocrine signaling, and epigenetic regulation. Understanding the underlying genetic defects in linear growth is important for clinicians and researchers in order to provide proper diagnoses, management, and genetic counseling, as well as to develop better treatment approaches for children with growth disorders.
MeSH Terms
Figure
-
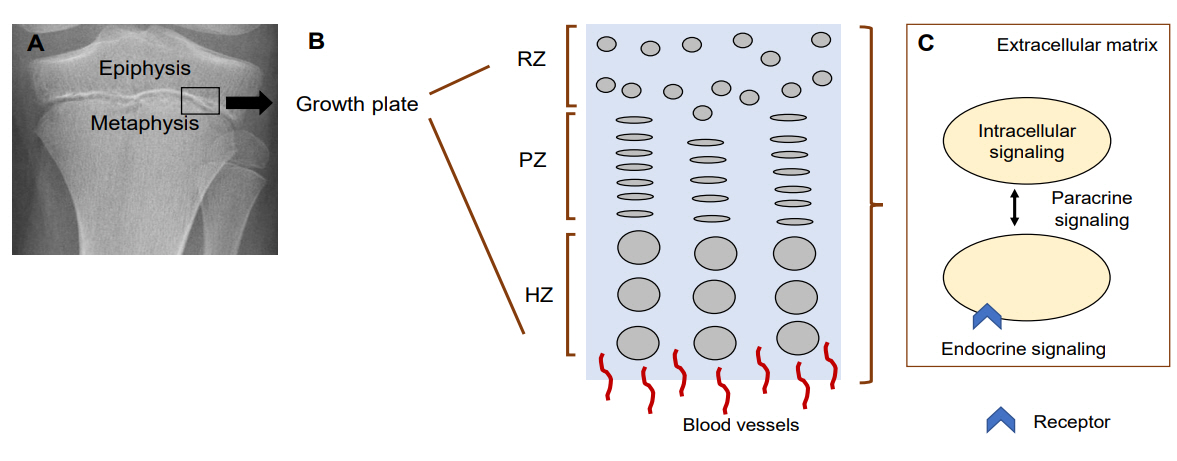
Fig. 1. Mechanisms regulating chondrocytes in the growth plate. (A) Growth plate on X-ray. Black box indicates where growth plate is located. (B) Diagram of the 3 layers in the growth plate. (C) Diagram of chondrocyte-regulating mechanisms. RZ, resting zone; PZ, proliferative zone; HZ, hypertrophic zone. Modified from Jee YH, et al. Endocrinol Metab Clin North Am 2017;46:259-81 [15].
Cited by 2 articles
-
Approach to Short Stature in Children and Adolescent
Hyo-Kyoung Nam
Ewha Med J. 2021;44(4):111-116. doi: 10.12771/emj.2021.44.4.111.Clinical validation of a deep-learning-based bone age software in healthy Korean children
Hyo-Kyoung Nam, Winnah Wu-In Lea, Zepa Yang, Eunjin Noh, Young-Jun Rhie, Kee-Hyoung Lee, Suk-Joo Hong
Ann Pediatr Endocrinol Metab. 2024;29(2):102-108. doi: 10.6065/apem.2346050.025.
Reference
-
References
1. Lango Allen H, Estrada K, Lettre G, Berndt SI, Weedon MN, Rivadeneira F, et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature. 2010; 467:832–8.2. Chan Y, Salem RM, Hsu YH, McMahon G, Pers TH, Vedantam S, et al. Genome-wide analysis of body proportion classifies height-associated variants by mechanism of action and implicates genes important for skeletal development. Am J Hum Genet. 2015; 96:695–708.
Article3. Marouli E, Graff M, Medina-Gomez C, Lo KS, Wood AR, Kjaer TR, et al. Rare and low-frequency coding variants alter human adult height. Nature. 2017; 542:186–90.4. Baron J, Sävendahl L, De Luca F, Dauber A, Phillip M, Wit JM, et al. Short and tall stature: a new paradigm emerges. Nat Rev Endocrinol. 2015; 11:735–46.
Article5. Lui JC, Nilsson O, Chan Y, Palmer CD, Andrade AC, Hirschhorn JN, et al. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height. Hum Mol Genet. 2012; 21:5193–201.
Article6. Tompson SW, Merriman B, Funari VA, Fresquet M, Lachman RS, Rimoin DL, et al. A recessive skeletal dysplasia, SEMD aggrecan type, results from a missense mutation affecting the C-type lectin domain of aggrecan. Am J Hum Genet. 2009; 84:72–9.
Article7. Nilsson O, Guo MH, Dunbar N, Popovic J, Flynn D, Jacobsen C, et al. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations. J Clin Endocrinol Metab. 2014; 99:E1510–8.
Article8. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003; 423:332–6.
Article9. Lui JC, Andrade AC, Forcinito P, Hegde A, Chen W, Baron J, et al. Spatial and temporal regulation of gene expression in the mammalian growth plate. Bone. 2010; 46:1380–90.
Article10. Melrose J, Shu C, Whitelock JM, Lord MS. The cartilage extracellular matrix as a transient developmental scaffold for growth plate maturation. Matrix Biol. 2016; 52-54:363–83.
Article11. Myllyharju J. Extracellular matrix and developing growth plate. Curr Osteoporos Rep. 2014; 12:439–45.
Article12. Cortes M, Baria AT, Schwartz NB. Sulfation of chondroitin sulfate proteoglycans is necessary for proper Indian hedgehog signaling in the developing growth plate. Development. 2009; 136:1697–706.
Article13. Lauing KL, Cortes M, Domowicz MS, Henry JG, Baria AT, Schwartz NB. Aggrecan is required for growth plate cytoarchitecture and differentiation. Dev Biol. 2014; 396:224–36.
Article14. Tatsi C, Gkourogianni A, Mohnike K, DeArment D, Witchel S, Andrade AC, et al. Aggrecan mutations in nonfamilial short stature and short stature without accelerated skeletal maturation. J Endocr Soc. 2017; 1:1006–11.
Article15. Gleghorn L, Ramesar R, Beighton P, Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis. Am J Hum Genet. 2005; 77:484–90.
Article16. Stattin EL, Wiklund F, Lindblom K, Onnerfjord P, Jonsson BA, Tegner Y, et al. A missense mutation in the aggrecan C-type lectin domain disrupts extracellular matrix interactions and causes dominant familial osteochondritis dissecans. Am J Hum Genet. 2010; 86:126–37.
Article17. Hauer NN, Sticht H, Boppudi S, Büttner C, Kraus C, Trautmann U, et al. Genetic screening confirms heterozygous mutations in ACAN as a major cause of idiopathic short stature. Sci Rep. 2017; 7:12225.18. Sakai LY, Keene DR. Fibrillin protein pleiotropy: Acromelic dysplasias. Matrix Biol. 2018; Sep. 13. [Epub]. pii: S0945-053X(18)30312-3. https://doi.org/10.1016/j.matbio.2018.09.005.
Article19. Haji-Seyed-Javadi R, Jelodari-Mamaghani S, Paylakhi SH, Yazdani S, Nilforushan N, Fan JB, et al. LTBP2 mutations cause Weill-Marchesani and Weill-Marchesani-like syndrome and affect disruptions in the extracellular matrix. Hum Mutat. 2012; 33:1182–7.
Article20. Kojuri J, Razeghinejad MR, Aslani A. Cardiac findings in Weill-Marchesani syndrome. Am J Med Genet A. 2007; 143A:2062–4.
Article21. Grau U, Klein HG, Detter C, Mair H, Welz A, Seidel D, et al. A novel mutation in the neonatal region of the fibrillin (FBN)1 gene associated with a classical phenotype of Marfan syndrome (MfS). Mutations in brief no. 163. Online. Hum Mutat. 1998; 12:137.
Article22. Cook JR, Ramirez F. Clinical, diagnostic, and therapeutic aspects of the Marfan syndrome. Adv Exp Med Biol. 2014; 802:77–94.
Article23. Park JW, Yan L, Stoddard C, Wang X, Yue Z, Crandall L, et al. Recapitulating and correcting Marfan syndrome in a cellular model. Int J Biol Sci. 2017; 13:588–603.
Article24. Cecchi A, Ogawa N, Martinez HR, Carlson A, Fan Y, Penny DJ, et al. Missense mutations in FBN1 exons 41 and 42 cause Weill-Marchesani syndrome with thoracic aortic disease and Marfan syndrome. Am J Med Genet A. 2013; 161A:2305–10.25. Putnam EA, Zhang H, Ramirez F, Milewicz DM. Fibrillin-2 (FBN2) mutations result in the Marfan-like disorder, congenital contractural arachnodactyly. Nat Genet. 1995; 11:456–8.
Article26. Malemud CJ. Matrix metalloproteinases: role in skeletal development and growth plate disorders. Front Biosci. 2006; 11:1702–15.
Article27. de Vos IJ, Tao EY, Ong SL, Goggi JL, Scerri T, Wilson GR, et al. Functional analysis of a hypomorphic allele shows that MMP14 catalytic activity is the prime determinant of the Winchester syndrome phenotype. Hum Mol Genet. 2018; 27:2775–88.
Article28. Shah MH, Bhat V, Shetty JS, Kumar A. Whole exome sequencing identifies a novel splice-site mutation in ADAMTS17 in an Indian family with Weill-Marchesani syndrome. Mol Vis. 2014; 20:790–6.29. Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrouqi RA, et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009; 85:558–68.
Article30. Hubmacher D, Apte SS. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. 2015; 47:34–43.
Article31. Zaucke F, Grässel S. Genetic mouse models for the functional analysis of the perifibrillar components collagen IX, COMP and matrilin-3: Implications for growth cartilage differentiation and endochondral ossification. Histol Histopathol. 2009; 24:1067–79.32. Plumb DA, Ferrara L, Torbica T, Knowles L, Mironov A Jr, Kadler KE, et al. Collagen XXVII organises the pericellular matrix in the growth plate. PLoS One. 2011; 6:e29422.
Article33. Hjorten R, Hansen U, Underwood RA, Telfer HE, Fernandes RJ, Krakow D, et al. Type XXVII collagen at the transition of cartilage to bone during skeletogenesis. Bone. 2007; 41:535–42.
Article34. Li Y, Lacerda DA, Warman ML, Beier DR, Yoshioka H, Ninomiya Y, et al. A fibrillar collagen gene, Col11a1, is essential for skeletal morphogenesis. Cell. 1995; 80:423–30.
Article35. Van Camp G, Snoeckx RL, Hilgert N, van den Ende J, Fukuoka H, Wagatsuma M, et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am J Hum Genet. 2006; 79:449–57.
Article36. Majava M, Hoornaert KP, Bartholdi D, Bouma MC, Bouman K, Carrera M, et al. A report on 10 new patients with heterozygous mutations in the COL11A1 gene and a review of genotype-phenotype correlations in type XI collagenopathies. Am J Med Genet A. 2007; 143A:258–64.37. Annunen S, Körkkö J, Czarny M, Warman ML, Brunner HG, Kääriäinen H, et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am J Hum Genet. 1999; 65:974–83.
Article38. Griffith AJ, Sprunger LK, Sirko-Osadsa DA, Tiller GE, Meisler MH, Warman ML. Marshall syndrome associated with a splicing defect at the COL11A1 locus. Am J Hum Genet. 1998; 62:816–23.
Article39. Tompson SW, Bacino CA, Safina NP, Bober MB, Proud VK, Funari T, et al. Fibrochondrogenesis results from mutations in the COL11A1 type XI collagen gene. Am J Hum Genet. 2010; 87:708–12.
Article40. Gariballa N, Ben-Mahmoud A, Komara M, Al-Shamsi AM, John A, Ali BR, et al. A novel aberrant splice site mutation in COL27A1 is responsible for Steel syndrome and extension of the phenotype to include hearing loss. Am J Med Genet A. 2017; 173:1257–63.41. Kotabagi S, Shah H, Shukla A, Girisha KM. Second family provides further evidence for causation of Steel syndrome by biallelic mutations in COL27A1. Clin Genet. 2017; 92:323–6.42. Gonzaga-Jauregui C, Gamble CN, Yuan B, Penney S, Jhangiani S, Muzny DM, et al. Mutations in COL27A1 cause Steel syndrome and suggest a founder mutation effect in the Puerto Rican population. Eur J Hum Genet. 2015; 23:342–6.
Article43. Jackson GC, Marcus-Soekarman D, Stolte-Dijkstra I, Verrips A, Taylor JA, Briggs MD. Type IX collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am J Med Genet A. 2010; 152A:863–9.
Article44. Baker S, Booth C, Fillman C, Shapiro M, Blair MP, Hyland JC, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A. 2011; 155A:1668–72.45. Mäkitie O, Mortier GR, Czarny-Ratajczak M, Wright MJ, Suri M, Rogala P, et al. Clinical and radiographic findings in multiple epiphyseal dysplasia caused by MATN3 mutations: description of 12 patients. Am J Med Genet A. 2004; 125A:278–84.
Article46. Baker S, Booth C, Fillman C, Shapiro M, Blair MP, Hyland JC, et al. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am J Med Genet A. 2011; 155A:1668–72.47. Borochowitz ZU, Scheffer D, Adir V, Dagoneau N, Munnich A, Cormier-Daire V. Spondylo-epi-metaphyseal dysplasia (SEMD) matrilin 3 type: homozygote matrilin 3 mutation in a novel form of SEMD. J Med Genet. 2004; 41:366–72.
Article48. Wagner T, Wirth J, Meyer J, Zabel B, Held M, Zimmer J, et al. Autosomal sex reversal and campomelic dysplasia are caused by mutations in and around the SRY-related gene SOX9. Cell. 1994; 79:1111–20.
Article49. Foster JW, Dominguez-Steglich MA, Guioli S, Kwok C, Weller PA, Stevanović M, et al. Campomelic dysplasia and autosomal sex reversal caused by mutations in an SRYrelated gene. Nature. 1994; 372:525–30.
Article50. Abou-Samra AB, Jüppner H, Force T, Freeman MW, Kong XF, Schipani E, et al. Expression cloning of a common receptor for parathyroid hormone and parathyroid hormone-related peptide from rat osteoblast-like cells: a single receptor stimulates intracellular accumulation of both cAMP and inositol trisphosphates and increases intracellular free calcium. Proc Natl Acad Sci U S A. 1992; 89:2732–6.
Article51. Singh AT, Gilchrist A, Voyno-Yasenetskaya T, Radeff-Huang JM, Stern PH. G alpha12/G alpha13 subunits of heterotrimeric G proteins mediate parathyroid hormone activation of phospholipase D in UMR-106 osteoblastic cells. Endocrinology. 2005; 146:2171–5.52. Turan S, Bastepe M. GNAS spectrum of disorders. Curr Osteoporos Rep. 2015; 13:146–58.
Article53. Mantovani G, Spada A. Mutations in the Gs alpha gene causing hormone resistance. Best Pract Res Clin Endocrinol Metab. 2006; 20:501–13.
Article54. Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007; 9:1–16.
Article55. Hennekam RC. Rubinstein-Taybi syndrome. Eur J Hum Genet. 2006; 14:981–5.
Article56. Maass PG, Aydin A, Luft FC, Schächterle C, Weise A, Stricker S, et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet. 2015; 47:647–53.
Article57. Boda H, Uchida H, Takaiso N, Ouchi Y, Fujita N, Kuno A, et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet. 2016; 61:701–3.
Article58. Vortkamp A, Lee K, Lanske B, Segre GV, Kronenberg HM, Tabin CJ. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science. 1996; 273:613–22.
Article59. Vasques GA, Funari MF, Ferreira FM, Aza-Carmona M, Sentchordi-Montané L, Barraza-García J, et al. IHH Gene Mutations causing short stature with nonspecific skeletal abnormalities and response to growth hormone therapy. J Clin Endocrinol Metab. 2018; 103:604–14.
Article60. Gao B, Guo J, She C, Shu A, Yang M, Tan Z, et al. Mutations in IHH, encoding Indian hedgehog, cause brachydactyly type A-1. Nat Genet. 2001; 28:386–8.
Article61. Hellemans J, Coucke PJ, Giedion A, De Paepe A, Kramer P, Beemer F, et al. Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips. Am J Hum Genet. 2003; 72:1040–6.
Article62. Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, et al. Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet. 2010; 86:434–9.
Article63. Zhang P, Jobert AS, Couvineau A, Silve C. A homozygous inactivating mutation in the parathyroid hormone/parathyroid hormone-related peptide receptor causing Blomstrand chondrodysplasia. J Clin Endocrinol Metab. 1998; 83:3365–8.
Article64. Schipani E, Kruse K, Jüppner H. A constitutively active mutant PTH-PTHrP receptor in Jansen-type metaphyseal chondrodysplasia. Science. 1995; 268:98–100.
Article65. Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006; 27:47–72.
Article66. Hisado-Oliva A, Ruzafa-Martin A, Sentchordi L, Funari MFA, Bezanilla-López C, Alonso-Bernáldez M, et al. Mutations in C-natriuretic peptide (NPPC): a novel cause of autosomal dominant short stature. Genet Med. 2018; 20:91–7.
Article67. Wang SR, Jacobsen CM, Carmichael H, Edmund AB, Robinson JW, Olney RC, et al. Heterozygous mutations in natriuretic peptide receptor-B (NPR2) gene as a cause of short stature. Hum Mutat. 2015; 36:474–81.68. Moncla A, Missirian C, Cacciagli P, Balzamo E, Legeai-Mallet L, Jouve JL, et al. A cluster of translocation breakpoints in 2q37 is associated with overexpression of NPPC in patients with a similar overgrowth phenotype. Hum Mutat. 2007; 28:1183–8.
Article69. Miura K, Kim OH, Lee HR, Namba N, Michigami T, Yoo WJ, et al. Overgrowth syndrome associated with a gainof-function mutation of the natriuretic peptide receptor 2 (NPR2) gene. Am J Med Genet A. 2014; 164A:156–63.70. Teixeira CC, Agoston H, Beier F. Nitric oxide, C-type natriuretic peptide and cGMP as regulators of endochondral ossification. Dev Biol. 2008; 319:171–8.71. Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. J Cell Sci. 2005; 118(Pt 21):5089–100.
Article72. Bartels CF, Bükülmez H, Padayatti P, Rhee DK, van Ravenswaaij-Arts C, Pauli RM, et al. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux. Am J Hum Genet. 2004; 75:27–34.73. Hachiya R, Ohashi Y, Kamei Y, Suganami T, Mochizuki H, Mitsui N, et sl. Intact kinase homology domain of natriuretic peptide receptor-B is essential for skeletal development. J Clin Endocrinol Metab. 2007; 92:4009–14.
Article74. Bocciardi R, Giorda R, Buttgereit J, Gimelli S, Divizia MT, Beri S, et al. Overexpression of the C-type natriuretic peptide (CNP) is associated with overgrowth and bone anomalies in an individual with balanced t(2;7) translocation. Hum Mutat. 2007; 28:724–31.
Article75. Miura K, Namba N, Fujiwara M, Ohata Y, Ishida H, Kitaoka T, et al. An overgrowth disorder associated with excessive production of cGMP due to a gain-of-function mutation of the natriuretic peptide receptor 2 gene. PLoS One. 2012; 7:e42180.
Article76. Hannema SE, van Duyvenvoorde HA, Premsler T, Yang RB, Mueller TD, Gassner B, et al. An activating mutation in the kinase homology domain of the natriuretic peptide receptor-2 causes extremely tall stature without skeletal deformities. J Clin Endocrinol Metab. 2013; 98:E1988–98.
Article77. Al-Qattan MM. Embryology of familial (non-syndromic) brachydactyly of the hand. J Hand Surg Eur Vol. 2014; 39:926–33.
Article78. Al-Qattan MM, Al-Motairi MI, Al Balwi MA. Two novel homozygous missense mutations in the GDF5 gene cause brachydactyly type C. Am J Med Genet A. 2015; 167:1621–6.79. Farooq M, Nakai H, Fujimoto A, Fujikawa H, Kjaer KW, Baig SM, et al. Characterization of a novel missense mutation in the prodomain of GDF5, which underlies brachydactyly type C and mild Grebe type chondrodysplasia in a large Pakistani family. Hum Genet. 2013; 132:1253–64.
Article80. Martinez-Garcia M, Garcia-Canto E, Fenollar-Cortes M, Aytes AP, Trujillo-Tiebas MJ. Characterization of an acromesomelic dysplasia, Grebe type case: novel mutation affecting the recognition motif at the processing site of GDF5. J Bone Miner Metab. 2016; 34:599–603.
Article81. Thomas JT, Kilpatrick MW, Lin K, Erlacher L, Lembessis P, Costa T, et al. Disruption of human limb morphogenesis by a dominant negative mutation in CDMP1. Nat Genet. 1997; 17:58–64.
Article82. Chang SC, Hoang B, Thomas JT, Vukicevic S, Luyten FP, Ryba NJ, et al. Cartilage-derived morphogenetic proteins. New members of the transforming growth factor-beta superfamily predominantly expressed in long bones during human embryonic development. J Biol Chem. 1994; 269:28227–34.
Article83. Hatakeyama Y, Tuan RS, Shum L. Distinct functions of BMP4 and GDF5 in the regulation of chondrogenesis. J Cell Biochem. 2004; 91:1204–17.
Article84. Bai X, Xiao Z, Pan Y, Hu J, Pohl J, Wen J, et al. Cartilagederived morphogenetic protein-1 promotes the differentiation of mesenchymal stem cells into chondrocytes. Biochem Biophys Res Commun. 2004; 325:453–60.
Article85. Kuss P, Kraft K, Stumm J, Ibrahim D, Vallecillo-Garcia P, Mundlos S, et al. Regulation of cell polarity in the cartilage growth plate and perichondrium of metacarpal elements by HOXD13 and WNT5A. Dev Biol. 2014; 385:83–93.
Article86. Person AD, Beiraghi S, Sieben CM, Hermanson S, Neumann AN, Robu ME, et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn. 2010; 239:327–37.87. Roifman M, Marcelis CL, Paton T, Marshall C, Silver R, Lohr JL, et al. De novo WNT5A-associated autosomal dominant Robinow syndrome suggests specificity of genotype and phenotype. Clin Genet. 2015; 87:34–41.
Article88. Uchimura T, Hollander JM, Nakamura DS, Liu Z, Rosen CJ, Georgakoudi I, et al. An essential role for IGF2 in cartilage development and glucose metabolism during postnatal long bone growth. Development. 2017; 144:3533–46.
Article89. Begemann M, Zirn B, Santen G, Wirthgen E, Soellner L, Büttel HM, et al. Paternally Inherited IGF2 Mutation and Growth Restriction. N Engl J Med. 2015; 373:349–56.
Article90. Nilsson O, Marino R, De Luca F, Phillip M, Baron J. Endocrine regulation of the growth plate. Horm Res. 2005; 64:157–65.
Article91. Daughaday WH. Growth hormone axis overview--somatomedin hypothesis. Pediatr Nephrol. 2000; 14:537–40.92. Fujimoto M, Kawashima Sonoyama Y, Hamajima N, Hamajima T, Kumura Y, Miyahara N, et al. Heterozygous nonsense mutations near the C-terminal region of IGF1R in two patients with small-for-gestational-age-related short stature. Clin Endocrinol (Oxf). 2015; 83:834–41.
Article93. Abuzzahab MJ, Schneider A, Goddard A, Grigorescu F, Lautier C, Keller E, et al. IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. N Engl J Med. 2003; 349:2211–22.
Article94. Dauber A, Muñoz-Calvo MT, Barrios V, Domené HM, Kloverpris S, Serra-Juhé C, et al. Mutations in pregnancyassociated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med. 2016; 8:363–74.
Article95. Bárcena C, Quesada V, De Sandre-Giovannoli A, Puente DA, Fernández-Toral J, Sigaudy S, et al. Exome sequencing identifies a novel mutation in PIK3R1 as the cause of SHORT syndrome. BMC Med Genet. 2014; 15:51.
Article96. Dyment DA, Smith AC, Alcantara D, Schwartzentruber JA, Basel-Vanagaite L, Curry CJ, et al. Mutations in PIK3R1 cause SHORT syndrome. Am J Hum Genet. 2013; 93:158–66.
Article97. Schroeder C, Riess A, Bonin M, Bauer P, Riess O, Döbler-Neumann M, et al. PIK3R1 mutations in SHORT syndrome. Clin Genet. 2014; 86:292–4.98. Nilsson O, Weise M, Landman EB, Meyers JL, Barnes KM, Baron J. Evidence that estrogen hastens epiphyseal fusion and cessation of longitudinal bone growth by irreversibly depleting the number of resting zone progenitor cells in female rabbits. Endocrinology. 2014; 155:2892–9.
Article99. Verma N, Jain V, Birla S, Jain R, Sharma A. Growth and hormonal profile from birth to adolescence of a girl with aromatase deficiency. J Pediatr Endocrinol Metab. 2012; 25:1185–90.
Article100. Marino R, Perez Garrido N, Costanzo M, Guercio G, Juanes M, Rocco C, et al. Five new cases of 46,XX aromatase deficiency: clinical follow-up from birth to puberty, a novel mutation, and a founder effect. J Clin Endocrinol Metab. 2015; 100:E301–7.
Article101. Fukami M, Miyado M, Nagasaki K, Shozu M, Ogata T. Aromatase excess syndrome: a rare autosomal dominant disorder leading to pre- or peri-pubertal onset gynecomastia. Pediatr Endocrinol Rev. 2014; 11:298–305.102. Quaynor SD, Stradtman EW Jr, Kim HG, Shen Y, Chorich LP, Schreihofer DA, et al. Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant. N Engl J Med. 2013; 369:164–71.
Article103. Bernard V, Kherra S, Francou B, Fagart J, Viengchareun S, Guéchot J, et al. Familial multiplicity of estrogen insensitivity associated with a loss-of-function ESR1 mutation. J Clin Endocrinol Metab. 2017; 102:93–9.104. Smith EP, Boyd J, Frank GR, Takahashi H, Cohen RM, Specker B, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994; 331:1056–61.
Article105. Eggermann T. Epigenetic regulation of growth: lessons from Silver-Russell syndrome. Endocr Dev. 2009; 14:10–9.
Article106. Tatton-Brown K, Seal S, Ruark E, Harmer J, Ramsay E, Del Vecchio Duarte S, et al. Mutations in the DNA methyltransferase gene DNMT3A cause an overgrowth syndrome with intellectual disability. Nat Genet. 2014; 46:385–8.
Article107. Xin B, Cruz Marino T, Szekely J, Leblanc J, Cechner K, Sency V, et al. Novel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome. Clin Genet. 2017; 91:623–8.108. Tatton-Brown K, Loveday C, Yost S, Clarke M, Ramsay E, Zachariou A, et al. Mutations in epigenetic regulation genes are a major cause of overgrowth with intellectual disability. Am J Hum Genet. 2017; 100:725–36.
Article109. Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, et al. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet. 2012; 90:110–8.
Article110. Tatton-Brown K, Hanks S, Ruark E, Zachariou A, Duarte Sdel V, Ramsay E, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget. 2011; 2:1127–33.
Article111. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039–43.
Article112. Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, et al. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 2002; 30:365–6.
Article113. Luscan A, Laurendeau I, Malan V, Francannet C, Odent S, Giuliano F, et al. Mutations in SETD2 cause a novel overgrowth condition. J Med Genet. 2014; 51:512–7.
Article114. Baujat G, Rio M, Rossignol S, Sanlaville D, Lyonnet S, Le Merrer M, et al. Paradoxical NSD1 mutations in Beckwith-Wiedemann syndrome and 11p15 anomalies in Sotos syndrome. Am J Hum Genet. 2004; 74:715–20.
Article115. Jee YH, Andrade AC, Baron J, Nilsson O. Genetics of short stature. Endocrinol Metab Clin North Am. 2017; 46:259–81.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Growth plate extracellular matrix defects and short stature in children
- Novel genetic cause of idiopathic short stature
- Approach to Short Stature in Children and Adolescent
- Recent Insights into Insulin-Like Growth Factor Binding Protein 2 Transcriptional Regulation
- A Functional Polymorphism in the DRD1 Gene, That Modulates Its Regulation by miR-504, Is Associated with Depressive Symptoms
