Lab Med Online.
2018 Jul;8(3):114-118. 10.3343/lmo.2018.8.3.114.
Two Korean Cases of Hereditary Spherocytosis Caused by Mutations in SLC4A1
- Affiliations
-
- 1Department of Laboratory Medicine, Seoul St. Mary's Hospital, Leukemia Research Institute, College of Medicine, The Catholic University of Korea, Seoul, Korea. yonggoo@catholic.ac.kr
- 2Catholic Genetic Laboratory Center, Seoul St. Mary's Hospital, Leukemia Research Institute, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 3Department of Pediatrics, College of Medicine, The Catholic University of Korea, Seoul St. Mary's Hospital, Leukemia Research Institute, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 4Department of Hematology, Seoul St. Mary's Hospital, Leukemia Research Institute, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- 5Catholic Blood and Marrow Transplantation Center, Seoul St. Mary's Hospital, Leukemia Research Institute, College of Medicine, The Catholic University of Korea, Seoul, Korea.
- KMID: 2414133
- DOI: http://doi.org/10.3343/lmo.2018.8.3.114
Abstract
- Hereditary spherocytosis (HS) is caused by mutations in the SPTA1, SPTB, ANK1, SLC4A1, and EPB42 genes, all of which encode erythrocyte membrane proteins. Mutations in SLC4A1, which encodes band 3 protein, have rarely been reported as the causative factor among Korean patients with HS. Here, we report two Korean patients with HS carrying mutations in SLC4A1. Patient 1 was a 3-year-old girl with unremarkable past and family histories and was evaluated for anemia that was detected after a complete blood count. She was suspected of having HS considering the spherocytosis of her peripheral blood smear, increased osmotic fragility, hemolytic features in blood chemistry tests, and splenomegaly. Sequence analysis revealed that the patient harbored a single heterozygous missense mutation, c.2278C>T (p.Arg760Trp) in exon 17 of SLC4A1. Patient 2 was a 23-year-old man who had a prior history of intermittent jaundice. Although the patient did not have anemia, a genetic test for HS was performed due to evidence of hemolytic features in the blood chemistry test, splenomegaly, and a family history of HS. The test confirmed a single heterozygous missense mutation, c.2423G>T (p.Arg808Leu) in exon 18 of SLC4A1.
Keyword
MeSH Terms
Figure
-
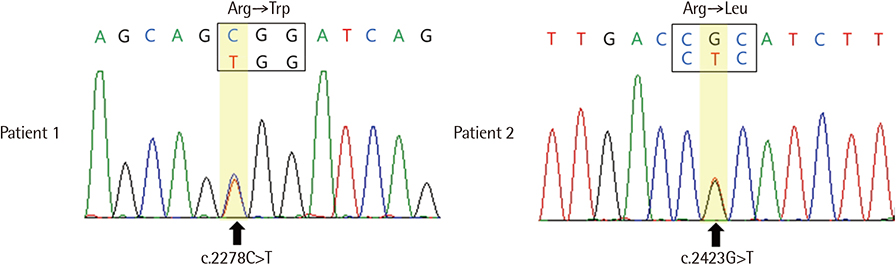
Fig. 1 Sequencing chromatograms of SLC4A1 mutations. (A) Patient 1, c.2278C>T (p.Arg760Trp) in exon 17; (B) Patient 2, c.2423G>T (p.Arg808Leu) in exon 18.
Reference
-
1. Eber S, Lux SE. Hereditary spherocytosis--defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004; 41:118–141.
Article2. Park J, Jeong DC, Yoo J, Jang W, Chae H, Kim J, et al. Mutational characteristics of ANK1 and SPTB genes in hereditary spherocytosis. Clin Genet. 2016; 90:69–78.
Article3. Eber SW, Gonzalez JM, Lux ML, Scarpa AL, Tse WT, Dornwell M, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996; 13:214–218.
Article4. Bouhassira EE, Schwartz RS, Yawata Y, Ata K, Kanzaki A, Qiu JJ, et al. An alanine-to-threonine substitution in protein 4.2 cDNA is associated with a Japanese form of hereditary hemolytic anemia (protein 4.2NIPPON). Blood. 1992; 79:1846–1854.
Article5. Miraglia del Giudice E, Nobili B, Francese M, D'Urso L, Iolascon A, Eber S, et al. Clinical and molecular evaluation of non-dominant hereditary spherocytosis. Br J Haematol. 2001; 112:42–47.
Article6. Bracher NA, Lyons CA, Wessels G, Mansvelt E, Coetzer TL. Band 3 Cape Town (E90K) causes severe hereditary spherocytosis in combination with band 3 Prague III. Br J Haematol. 2001; 113:689–693.
Article7. An X, Mohandas N. Disorders of red cell membrane. Br J Haematol. 2008; 141:367–375.
Article8. Choi K. Molecular diagnosis of hereditary spherocytosis by multi-gene target sequencing in Korea (Doctoral dissertation). Seoul, Korea: Seoul National University College of Medicine;2016.9. Rosenberg HK, Markowitz RI, Kolberg H, Park C, Hubbard A, Bellah RD. Normal splenic size in infants and children: sonographic measurements. AJR Am J Roentgenol. 1991; 157:119–121.
Article10. Jarolim P, Rubin HL, Brabec V, Chrobak L, Zolotarev AS, Alper SL, et al. Mutations of conserved arginines in the membrane domain of erythroid band 3 lead to a decrease in membrane-associated band 3 and to the phenotype of hereditary spherocytosis. Blood. 1995; 85:634–640.
Article11. Yawata Y, Kanzaki A, Yawata A, Doerfler W, Ozcan R, Eber SW. Characteristic features of the genotype and phenotype of hereditary spherocytosis in the Japanese population. Int J Hematol. 2000; 71:118–135.12. Quilty JA, Reithmeier RA. Trafficking and folding defects in hereditary spherocytosis mutants of the human red cell anion exchanger. Traffic. 2000; 1:987–998.
Article13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424.
Article14. Bogardus HH, Maksimova YD, Forget BG, Gallagher PG. A de novo band 3 mutation in hereditary spherocytosis. Pediatr Blood Cancer. 2012; 58:1004.
Article15. Da Costa L, Galimand J, Fenneteau O, Mohandas N. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013; 27:167–178.
Article16. Alloisio N, Texier P, Vallier A, Ribeiro ML, Morle L, Bozon M, et al. Modulation of clinical expression and band 3 deficiency in hereditary spherocytosis. Blood. 1997; 90:414–420.
Article17. Toye AM, Banting G, Tanner MJ. Regions of human kidney anion exchanger 1 (kAE1) required for basolateral targeting of kAE1 in polarised kidney cells: mis-targeting explains dominant renal tubular acidosis (dRTA). J Cell Sci. 2004; 117:1399–1410.
Article18. Lima PR, Gontijo JA, Lopes de Faria JB, Costa FF, Saad ST. Band 3 Campinas: a novel splicing mutation in the band 3 gene (AE1) associated with hereditary spherocytosis, hyperactivity of Na+/Li+ countertransport and an abnormal renal bicarbonate handling. Blood. 1997; 90:2810–2818.
Article19. Jarolim P, Palek J, Amato D, Hassan K, Sapak P, Nurse GT, et al. Deletion in erythrocyte band 3 gene in malaria-resistant Southeast Asian ovalocytosis. Proc Natl Acad Sci U S A. 1991; 88:11022–11026.
Article20. Bruce LJ, Kay MM, Lawrence C, Tanner MJ. Band 3 HT, a human red-cell variant associated with acanthocytosis and increased anion transport, carries the mutation Pro-868 → Leu in the membrane domain of band 3. Biochem J. 1993; 293:317–320.
Article21. Jarolim P, Rubin HL, Zhai S, Sahr KE, Liu SC, Mueller TJ, et al. Band 3 Memphis: a widespread polymorphism with abnormal electrophoretic mobility of erythrocyte band 3 protein caused by substitution AAG → GAG (Lys → Glu) in codon 56. Blood. 1992; 80:1592–1598.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Identification of a de novo ANK1 Variant in a Patient with Hereditary Spherocytosis on Multi-gene Panel Testing
- Serial CT Findings of Resolving Extramedullary Hematopoiesis as Unilateral Posterior Mediastinal Mass after Splenectomy in Hereditary Spherocytosis: A Case Report
- Hereditary Spherocytosis
- Cholangitis Caused by Hereditary Spherocytosis in Adulthood Treated by Endoscopic Retrograde Cholangiopancreatography
- Flow-Assisted Differential Diagnosis of Hemolytic Anemia with Spherocytosis: A Case Report
