Enhancing T Cell Immune Responses by B Cell-based Therapeutic Vaccine Against Chronic Virus Infection
- Affiliations
-
- 1System Immunology Laboratory, Department of Biochemistry, College of Life Science and Biotechnology, Yonsei University, Seoul 120-749, Korea. sjha@yonsei.ac.kr
- 2Cell Therapy Team, Mogam Biotechnology Institute, Yongin 446-799, Korea.
- 3College of Pharmacy, Seoul National University, Seoul 110-799, Korea.
- KMID: 2150809
- DOI: http://doi.org/10.4110/in.2014.14.4.207
Abstract
- Chronic virus infection leads to the functional impairment of dendritic cells (DCs) as well as T cells, limiting the clinical usefulness of DC-based therapeutic vaccine against chronic virus infection. Meanwhile, B cells have been known to maintain the ability to differentiate plasma cells producing antibodies even during chronic virus infection. Previously, alpha-galactosylceramide (alphaGC) and cognate peptide-loaded B cells were comparable to DCs in priming peptide-specific CD8+ T cells as antigen presenting cells (APCs). Here, we investigated whether B cells activated by alphaGC can improve virus-specific T cell immune responses instead of DCs during chronic virus infection. We found that comparable to B cells isolated from naive mice, chronic B cells isolated from chronically infected mice with lymphocytic choriomeningitis virus (LCMV) clone 13 (CL13) after alphaGC-loading could activate CD1d-restricted invariant natural killer T (iNKT) cells to produce effector cytokines and upregulate co-stimulatory molecules in both naive and chronically infected mice. Similar to naive B cells, chronic B cells efficiently primed LCMV glycoprotein (GP) 33-41-specific P14 CD8+ T cells in vivo, thereby allowing the proliferation of functional CD8+ T cells. Importantly, when alphaGC and cognate epitope-loaded chronic B cells were transferred into chronically infected mice, the mice showed a significant increase in the population of epitope-specific CD8+ T cells and the accelerated control of viremia. Therefore, our studies demonstrate that reciprocal activation between alphaGC-loaded chronic B cells and iNKT cells can strengthen virus-specific T cell immune responses, providing an effective regimen of autologous B cell-based therapeutic vaccine to treat chronic virus infection.
Keyword
MeSH Terms
Figure
-

Figure 1 Comparison of αGC-loaded naïve or chronic B cells for bidirectional activation of iNKT and B cells in naïve mice. naïve and chronic B cells were isolated from splenocytes of naïve mice and chronically infected mice that were initially depleted of CD4+ T cells and subsequently infected with LCMV CL13 (over 90 d p.i.), respectively. (A) Schedule for αGC-loading onto B cells and generation of in vivo activated iNKT cells and B cells. Naive and chronic B cells isolated from Ly5.1+ naïve and chronically infected mice were cultured with vehicle (veh) or αGC in vitro for 18~20 h. 2×106 cells of αGC-loaded B cells were adoptively transferred into Ly5.2+ congenic naive mice. The recipient mice were sacrificed 6 and 24 h after adoptive transfer of B cells for analysis of the activation of iNKT cells and donor B cells, respectively. (B) In vivo activation of iNKT cells by αGC-loaded naïve and chronic B cells in naïve mice. Frequency of iNKT cells among CD4+ T cells was examined by staining with CD1d tetramer (tet) and TCR-β (CD1d tet+ TCR-βinter) and their function was evaluated by intracellular staining of IFN-γ and TNF-α. The number in the plot indicates the percent of corresponding population. (C) In vivo activation of donor αGC-loaded B cells by activated iNKT cells in naïve mice. Ly5.1+ CD19+ donor B cells were gated and analyzed for the expression of surface molecules. The number in histogram plot represents mean fluorescence intensity (MFI) of the expressed protein. The vertical grey line in histogram plot indicate a geometric mean level of the protein expressed on naïve B cells loaded with vehicle. Results are representative of at least three independent experiments.

Figure 2 Comparison of αGC-loaded naïve or chronic B cells for bidirectional activation of iNKT and B cells in chronically infected mice. Naïve and chronic B cells were isolated from splenocytes of naïve mice and chronically infected mice that were initially depleted of CD4+ T cells and subsequently infected with LCMV CL13 (over 90 d p.i.), respectively. (A) Schedule for αGC-loading onto B cells and generation of in vivo activated iNKT cells and B cells. Naive and chronic B cells isolated from Ly5.1+ naïve and chronically infected mice were cultured with veh or αGC in vitro for 18~20 h. 2×106 cells of αGC-loaded B cells were adoptively transferred into Ly5.2+ congenic mice that were already infected with LCMV CL13 (over 90 d p.i.). The recipient mice were sacrificed 6 and 24 h after adoptive transfer of B cells for analysis of the activation of iNKT cells and donor B cells, respectively. (B) In vivo activation of iNKT cells by αGC-loaded naïve and chronic B cells in chronically infected mice. Frequency of iNKT cells among CD4+ T cells was examined by staining with CD1d tet and TCR-β (CD1d tet+ TCR-βinter) and their function was evaluated by intracellular staining of IFN-γ and TNF-α. The number in the plot indicates the percent of corresponding population. (C) In vivo activation of donor αGC-loaded B cells by activated iNKT cells in chronically infected mice. Ly5.1+ CD19+ donor B cells were gated and analyzed for the expression of surface molecules. The number in histogram plot represents mean fluorescence intensity (MFI) of the expressed protein. The vertical grey line in histogram plot indicate a geometric mean level of the protein expressed on naïve B cells loaded with vehicle. Results are representative of at least three independent experiments.

Figure 3 In vivo priming of antigen-specific CD8+ T cells by αGC and epitope-loaded naïve and chronic B cells. Naïve and chronic B cells were isolated from splenocytes of naïve mice and chronically infected mice that were initially depleted of CD4+ T cells and subsequently infected with LCMV CL13 (over 90 d p.i.), respectively. Naïve P14 CD8+ T cells were purified from splenocytes of naïve P14 mice. (A) Schedule for analysis for in vivo activity of αGC and peptide-loaded naïve and chronic B cells to prime antigen-specific CD8+ T cells. 5×106 of CellTrace™ Violet (CTV)-labeled P14 Thy1.1+ CD8+ T cells were adoptively transferred into Thy1.2+ congenic naïve mice. After 24 h, the mice were given with 1×106 of naïve or chronic B cells loaded with veh, αGC, veh plus GP33-41 peptide (GP33), or αGC plus GP33. The recipient mice were sacrificed 48 h after adoptive transfer of B cells for analysis of donor P14 cells. (B and C) Proliferation (1st row) and cytokine production (2nd to 4th row) of P14 cells primed with αGC plus GP33-loaded naïve (B) and chronic B cells (C). Donor Thy1.1+ CD8+ T cells in the spleen were gated and examined for CTV dilution along with cytokine production. Division time and frequency of dividing cell population are indicated in histogram plot. The number as shown in each quadrant of the plot represents percentage of the corresponding cell population. Results are representative of at least three independent experiments.

Figure 4 Effect of therapeutic vaccination with αGC and epitope-loaded naïve B cells on T cell responses and virus control during chronic virus infection. LCMV CL13-infected mice were vaccinated with 1×107 of veh, αGC, veh plus GP33, or αGC plus GP33-loaded naïve B cells at 25 d p.i. (A) Frequency of DbGP33-41 tet-positive cells among CD8+ T cells in the chronically infected mice after therapeutic vaccination with B cells. At 0, 7, and 14 d post-vaccination (25, 32, and 39 d p.i.), CD8+ T cells in the blood were gated and evaluated for the expansion of DbGP33-41 tet+ cells. (B) Fold-increase of DbGP33-41 tetramer specific CD8+ T cells in PBMCs between 0 and 7 d post-vaccination. (C) Change of virus titer in the blood post-vaccination. Serum virus titer was determined from individual mice before (17 d p.i.) and after therapeutic vaccination with B cells (39 d p.i.). Vertical line in the graph indicates time point when therapeutic vaccination was performed. (D) Reduction of serum virus titer after B cell therapeutic vaccination. Fold-decrease of serum virus titer was calculated by serum virus titer before therapeutic vaccination (17 d p.i.) divided by that after therapeutic vaccination with B cells (39 d p.i.). Results are representative of two independent experiments. n=4 mice per group in each experiment. ns, not significant; *p<0.05; **p<0.01.
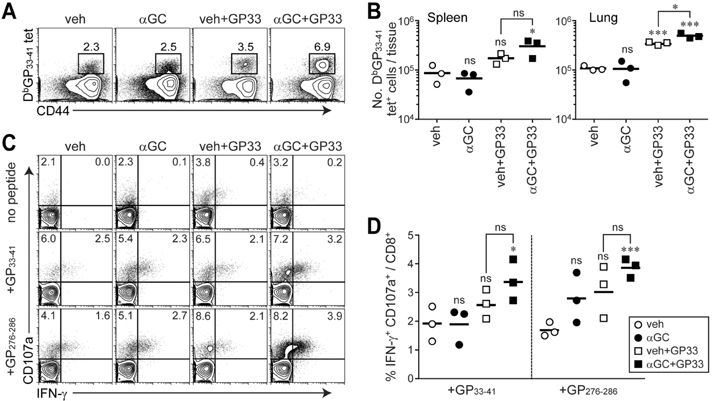
Figure 5 Expansion of epitope-specific CD8+ T cells and their effector function after therapeutic vaccination with αGC and epitope-loaded naïve B cells. LCMV CL13-infected mice were vaccinated with 1×107 of veh, αGC, veh plus GP33, or αGC plus GP33-loaded naïve B cells at 25 d p.i. and sacrificed at 39 d p.i. (14 d post-vaccination). (A) Frequency of DbGP33-41 tet+ cells among CD8+ T cells in the spleen. (B) Total number of DbGP33-41 tet+ cells in the spleen and lung. (C) IFN-γ and CD107a expression in CD8+ T cells in the spleen after in vitro stimulation with GP33-41 or GP276-286 peptide. (D) Proportion of CD8+ T cells producing IFN-γ and CD107a in the spleen after peptide stimulation. Number in the plot indicates percentage of the corresponding cells. Results are representative of two independent experiments. n=3 mice per group in each experiment. ns, not significant; *p<0.05; ***p<0.001.
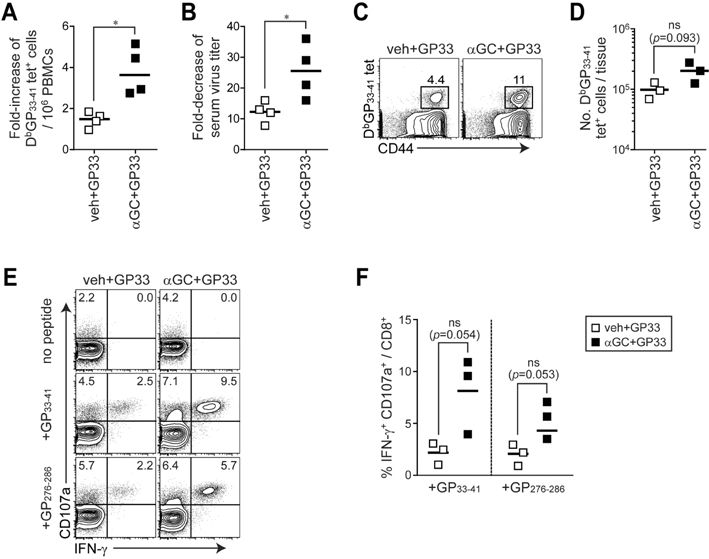
Figure 6 Therapeutic efficacy of αGC and epitope-loaded chronic B cells during chronic virus infection. Four different groups of B cells were purified from LCMV CL13-infected mice (25 d p.i.) and treated with veh plus GP33 or αGC plus GP33. LCMV CL13-infected mice were vaccinated at 25 d p.i. with 1×107 of each group of B cells as therapeutic vaccine and sacrificed at 39 d p.i. (14 d post-vaccination). (A) Fold-increase of DbGP33-41 tetramer specific CD8+ T cells in PBMCs between 0 and 7 d post-vaccination (between 25 and 32 d p.i.),. (B) Reduction of serum virus titer after B cell therapeutic vaccination. Fold-decrease of serum virus titer was calculated by serum virus titer before therapeutic vaccination (17 d p.i.) divided by that after therapeutic vaccination with B cells (39 d p.i.). (C) Frequency of DbGP33-41 tet+ cells among CD8+ T cells in the spleen. (D) Total number of DbGP33-41 tet+ cells in the spleen. (E) IFN-γ and CD107a expression in CD8+ T cells in the spleen after in vitro stimulation with GP33-41 or GP276-286 peptide. (F) Proportion of CD8+ T cells producing IFN-γ and CD107a in the spleen after peptide stimulation. Number in the plot indicates percentage of the corresponding cells. Results are representative of two independent experiments. n=3 mice per group in each experiment. ns, not significant.
Reference
-
1. Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J Virol. 2003; 77:4911–4927.
Article2. Ha SJ, West EE, Araki K, Smith KA, Ahmed R. Manipulating both the inhibitory and stimulatory immune system towards the success of therapeutic vaccination against chronic viral infections. Immunol Rev. 2008; 223:317–333.
Article3. Virgin HW, Wherry EJ, Ahmed R. Redefining chronic viral infection. Cell. 2009; 138:30–50.
Article4. Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J Exp Med. 1998; 188:2205–2213.
Article5. Jin HT, Jeong YH, Park HJ, Ha SJ. Mechanism of T cell exhaustion in a chronic environment. BMB Rep. 2011; 44:217–231.
Article6. Ng CT, Snell LM, Brooks DG, Oldstone MB. Networking at the level of host immunity: immune cell interactions during persistent viral infections. Cell Host Microbe. 2013; 13:652–664.
Article7. Fahey LM, Wilson EB, Elsaesser H, Fistonich CD, McGavern DB, Brooks DG. Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J Exp Med. 2011; 208:987–999.
Article8. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004; 10:909–915.
Article9. Figdor CG, de Vries IJ, Lesterhuis WJ, Melief CJ. Dendritic Cell Immunotherapy: Mapping The Way. Nat Med. 2004; 10:475–480.
Article10. Janikashvili N, Larmonier N, Katsanis E. Personalized dendritic cell-based tumor immunotherapy. Immunotherapy. 2010; 2:57–68.
Article11. Schultze JL, Grabbe S, von Bergwelt-Baildon MS. DCs and CD40-activated B cells: current and future avenues to cellular cancer immunotherapy. Trends Immunol. 2004; 25:659–664.
Article12. Eynon EE, Parker DC. Small B cells as antigen-presenting cells in the induction of tolerance to soluble protein antigens. J Exp Med. 1992; 175:131–138.
Article13. Fuchs EJ, Matzinger P. B cells turn off virgin but not memory T cells. Science. 1992; 258:1156–1159.
Article14. Schultze JL, Michalak S, Seamon MJ, Dranoff G, Jung K, Daley J, Delgado JC, Gribben JG, Nadler LM. CD40-activated human B cells: an alternative source of highly efficient antigen presenting cells to generate autologous antigen-specific T cells for adoptive immunotherapy. J Clin Invest. 1997; 100:2757–2765.
Article15. Coughlin CM, Vance BA, Grupp SA, Vonderheide RH. RNA-transfected CD40-activated B cells induce functional T-cell responses against viral and tumor antigen targets: implications for pediatric immunotherapy. Blood. 2004; 103:2046–2054.
Article16. Lapointe R, Bellemare-Pelletier A, Housseau F, Thibodeau J, Hwu P. CD40-stimulated B lymphocytes pulsed with tumor antigens are effective antigen-presenting cells that can generate specific T cells. Cancer Res. 2003; 63:2836–2843.17. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol. 2007; 25:297–336.
Article18. Godfrey DI, Stankovic S, Baxter AG. Raising the NKT cell family. Nat Immunol. 2010; 11:197–206.
Article19. Kronenberg M. Toward an understanding of NKT cell biology: progress and paradoxes. Annu Rev Immunol. 2005; 23:877–900.
Article20. Park SH, Bendelac A. CD1-restricted T-cell responses and microbial infection. Nature. 2000; 406:788–792.
Article21. Chung Y, Kim BS, Kim YJ, Ko HJ, Ko SY, Kim DH, Kang CY. CD1d-restricted T cells license B cells to generate long-lasting cytotoxic antitumor immunity in vivo. Cancer Res. 2006; 66:6843–6850.
Article22. Godfrey DI, Kronenberg M. Going both ways: immune regulation via CD1d-dependent NKT cells. J Clin Invest. 2004; 114:1379–1388.
Article23. Kim YJ, Ko HJ, Kim YS, Kim DH, Kang S, Kim JM, Chung Y, Kang CY. alpha-Galactosylceramide-loaded, antigen-expressing B cells prime a wide spectrum of antitumor immunity. Int J Cancer. 2008; 122:2774–2783.
Article24. Hermans IF, Silk JD, Gileadi U, Salio M, Mathew B, Ritter G, Schmidt R, Harris AL, Old L, Cerundolo V. NKT cells enhance CD4+ and CD8+ T cell responses to soluble antigen in vivo through direct interaction with dendritic cells. J Immunol. 2003; 171:5140–5147.
Article25. Stober D, Jomantaite I, Schirmbeck R, Reimann J. NKT cells provide help for dendritic cell-dependent priming of MHC class I-restricted CD8+ T cells in vivo. J Immunol. 2003; 170:2540–2548.
Article26. Cerundolo V, Silk JD, Masri SH, Salio M. Harnessing invariant NKT cells in vaccination strategies. Nat Rev Immunol. 2009; 9:28–38.
Article27. Ha SJ, Mueller SN, Wherry EJ, Barber DL, Aubert RD, Sharpe AH, Freeman GJ, Ahmed R. Enhancing therapeutic vaccination by blocking PD-1-mediated inhibitory signals during chronic infection. J Exp Med. 2008; 205:543–555.
Article28. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006; 439:682–687.
Article29. King CC, de Fries R, Kolhekar SR, Ahmed R. In vivo selection of lymphocyte-tropic and macrophage-tropic variants of lymphocytic choriomeningitis virus during persistent infection. J Virol. 1990; 64:5611–5616.
Article30. Elsaesser H, Sauer K, Brooks DG. IL-21 is required to control chronic viral infection. Science. 2009; 324:1569–1572.
Article31. Frohlich A, Kisielow J, Schmitz I, Freigang S, Shamshiev AT, Weber J, Marsland BJ, Oxenius A, Kopf M. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science. 2009; 324:1576–1580.
Article32. Yi JS, Du M, Zajac AJ. A vital role for interleukin-21 in the control of a chronic viral infection. Science. 2009; 324:1572–1576.
Article33. Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG. IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med. 2010; 207:353–363.
Article34. Zotos D, Coquet JM, Zhang Y, Light A, D'Costa K, Kallies A, Corcoran LM, Godfrey DI, Toellner KM, Smyth MJ, Nutt SL, Tarlinton DM. IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J Exp Med. 2010; 207:365–378.
Article35. Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat Rev Immunol. 2002; 2:116–126.
Article36. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, Ahmed R. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007; 27:670–684.
Article37. Wiesner M, Zentz C, Mayr C, Wimmer R, Hammerschmidt W, Zeidler R, Moosmann A. Conditional immortalization of human B cells by CD40 ligation. PLoS One. 2008; 3:e1464.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Porcine reproductive and respiratory syndrome virus vaccine does not fit in classical vaccinology
- Baculovirus-based Vaccine Displaying Respiratory Syncytial Virus Glycoprotein Induces Protective Immunity against RSV Infection without Vaccine-Enhanced Disease
- Enhancement of DNA Vaccine-induced Immune Responses by Influenza Virus NP Gene
- Vaccine Strategy That Enhances the Protective Efficacy of Systemic Immunization by Establishing LungResident Memory CD8 T Cells Against Influenza Infection
- The Role of CD4 T Cell Help in CD8 T Cell Differentiation and Function During Chronic Infection and Cancer
