Identification of a Novel Deletion Region in 3q29 Microdeletion Syndrome by Oligonucleotide Array Comparative Genomic Hybridization
- Affiliations
-
- 1Department of Laboratory Medicine, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea. ejseo@amc.seoul.kr
- 2Department of Pediatrics, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea.
- 3Department of Psychiatry, University of Ulsan College of Medicine and Asan Medical Center, Seoul, Korea.
- 4Asan Institute for Life Sciences, Seoul, Korea.
- KMID: 1096822
- DOI: http://doi.org/10.3343/kjlm.2010.30.1.70
Abstract
- BACKGROUND
The 3q29 microdeletion syndrome is a genomic disorder characterized by mental retardation, developmental delay, microcephaly, and slight facial dysmorphism. In most cases, the microdeletion spans a 1.6-Mb region between low-copy repeats (LCRs). We identified a novel 4.0- Mb deletion using oligonucleotide array comparative genomic hybridization (array CGH) in monozygotic twin sisters.
METHODS
G-banded chromosome analysis was performed in the twins and their parents. Highresolution oligonucleotide array CGH was performed using the human whole genome 244K CGH microarray (Agilent Technologies, USA) followed by validation using FISH, and the obtained results were analyzed using the genome database resources.
RESULTS
G-banding revealed that the twins had de novo 46,XX,del(3)(q29) karyotype. Array CGH showed a 4.0-Mb interstitial deletion on 3q29, which contained 39 genes and no breakpoints flanked by LCRs. In addition to the typical characteristics of the 3q29 microdeletion syndrome, the twins had attention deficit-hyperactivity disorder, strabismus, congenital heart defect, and gray hair. Besides the p21-activated protein kinase (PAK2) and discs large homolog 1 (DLG1) genes, which are known to play a critical role in mental retardation, the hairy and enhancer of split 1 (HES1) and antigen p97 (melanoma associated; MFI2) genes might be possible candidate genes associated with strabismus, congenital heart defect, and gray hair.
CONCLUSIONS
The novel 4.0-Mb 3q29 microdeletion found in the twins suggested the occurrence of genomic rearrangement mediated by mechanisms other than nonallelic homologous recombination. Molecular genetic and functional studies are required to elucidate the contribution of each gene to a specific phenotype.
Keyword
MeSH Terms
-
Adaptor Proteins, Signal Transducing/genetics
Adolescent
Attention Deficit Disorder with Hyperactivity/genetics
Basic Helix-Loop-Helix Transcription Factors/genetics
*Chromosome Deletion
Chromosome Disorders/*genetics
*Chromosomes, Human, Pair 3
Comparative Genomic Hybridization/*methods
Diseases in Twins/*genetics
Female
Homeodomain Proteins/genetics
Humans
In Situ Hybridization, Fluorescence
Melanoma-Specific Antigens/genetics
Membrane Proteins/genetics
Oligonucleotide Array Sequence Analysis
Syndrome
Twins
p21-Activated Kinases/genetics
Figure
-
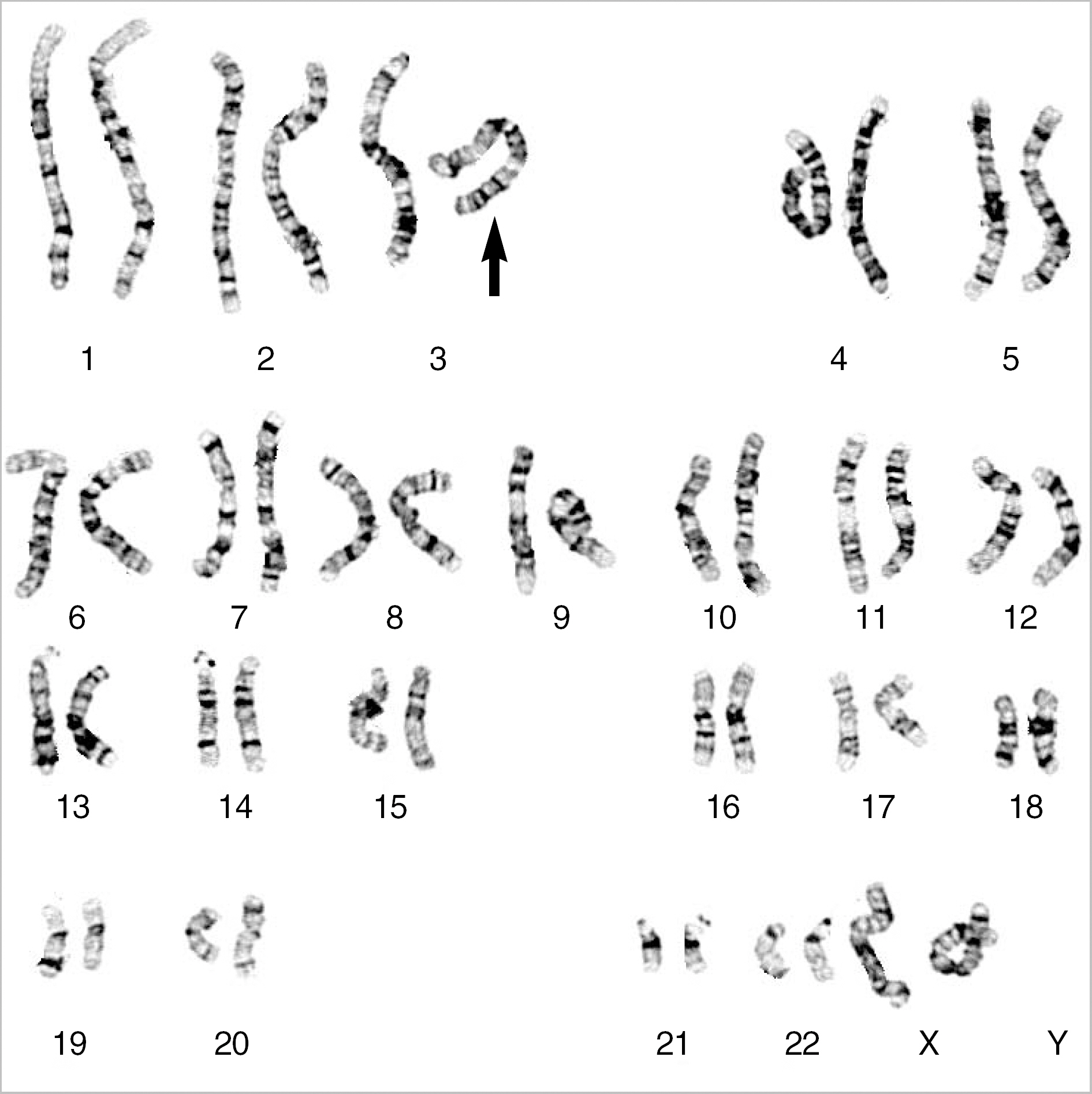
Fig. 1. Karyotype of one of the monozygotic twin sisters. The deletion in the long arm of chromosome 3 was apparently a terminal deletion, as shown by the arrow. The karyotype was initially designated 46,XX,del(3)(q29).
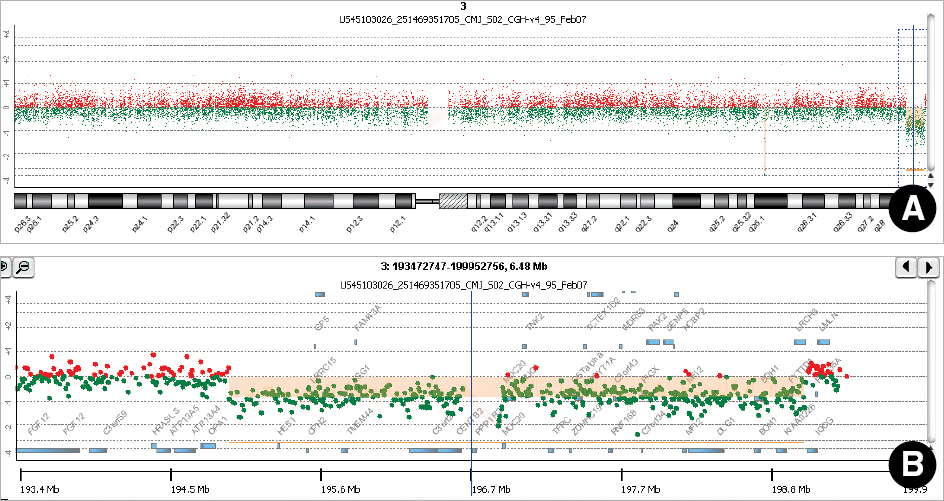
Fig. 2. Array CGH profile of chromosome 3 in one of the monozygotic twin sisters. X-axis represents the probe index on chromosome 3, and Y-axis represents the signal log2 ratio of the probe. (A) The whole chromosome 3 view showed copy number loss in the 3q29 region. The green dots with log2 value of −1 represent a 1:2 copy number ratio of the test to reference genomic DNA, indicating a heterozygous deletion. (B) The expansion view of the 3q29 region distinctly revealed a 4.0-Mb heterozygous interstitial deletion in the Chr3:195,007,970-199,085,431.

Fig. 3. Metaphase fluorescence in situ hybridization (FISH) analysis of one of the monozygotic twin sisters using TelVysion 3q SpectrumOrange probe and TelVysion 3p SpectrumGreen probe (Abbott Molecular Inc., USA). (A) TelVysion 3q deletion was detected (arrow), but 2 TelVysion 3p signals were intact. (B) TelVysion 3q deletion was also identified in the same metaphase with G-banding obtained using 4,6-diamidino-2-phenylindole (DAPI) staining (arrow).
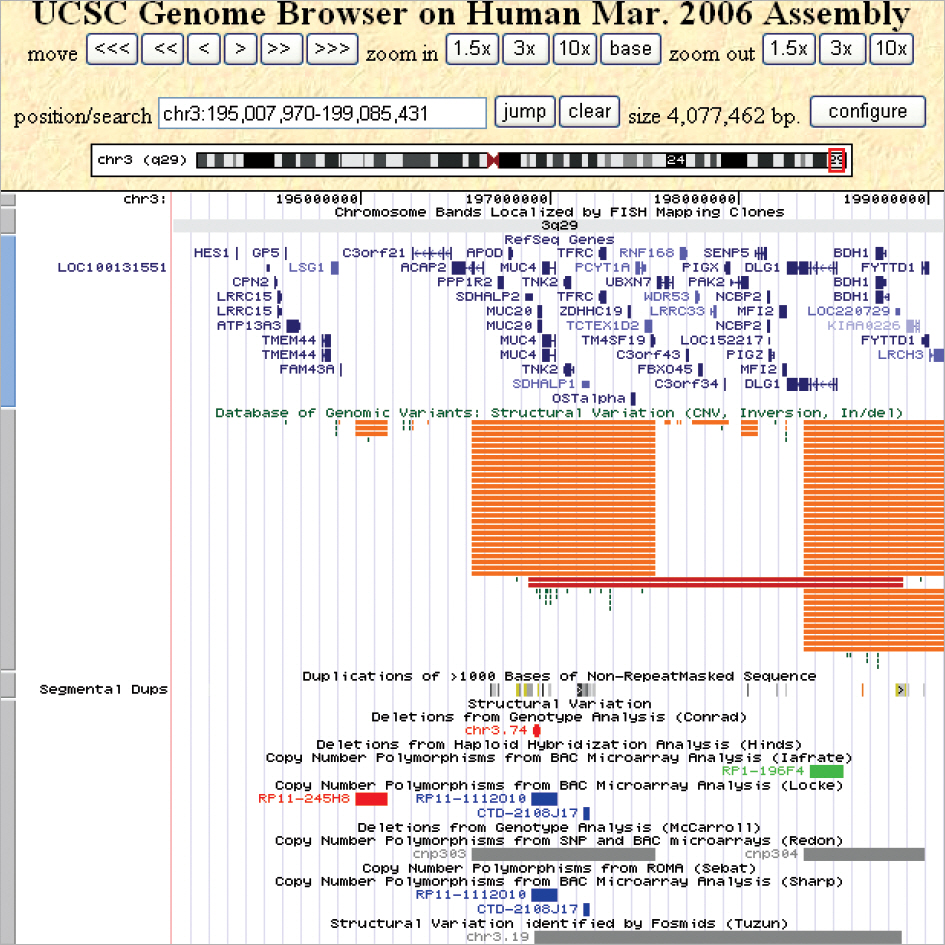
Fig. 4. Visualization of the novel deletion region on 3q29 using the University of California Santa Cruz (UCSC) Genome Browser. The UCSC browser shows a 4.0-Mb region (within positions Chr3:195,007,970-199,085,431), encompassing the reference genes, copy number variation regions, and segmental duplications.
Reference
-
1.Rossi E., Piccini F., Zollino M., Neri G., Caselli D., Tenconi R, et al. Cryptic telomeric rearrangements in subjects with mental retardation associated with dysmorphism and congenital malformations. J Med Genet. 2001. 38:417–20.
Article2.Willatt L., Cox J., Barber J., Cabanas ED., Collins A., Donnai D, et al. 3q29 microdeletion syndrome: clinical and molecular characterization of a new syndrome. Am J Hum Genet. 2005. 77:154–60.
Article3.Ballif BC., Theisen A., Coppinger J., Gowans GC., Hersh JH., Madan-Khetarpal S, et al. Expanding the clinical phenotype of the 3q29 microdeletion syndrome and characterization of the reciprocal micro-duplication. Mol Cytogenet. 2008. 1:8.
Article4.Baynam G., Goldblatt J., Townshend S. A case of 3q29 microdeletion with novel features and a review of cytogenetically visible terminal 3q deletions. Clin Dysmorphol. 2006. 15:145–8.
Article5.Genome Bioinformatics Group of UC Santa Cruz, UCSC genome browser. http://genome.ucsc.edu/cgi-bin/hgGateway. (Updated on Jan. 2009.6.The Centre for Applied Genomics, Database of genomic variants. http://projects.tcag.ca/variation/?source=hg18. (Updated on Jan. 2009.7.Online Mendelian Inheritance in Man. http://www.ncbi.nlm.nih.gov/sites/entrez?db=omim. (Accessed on Jan. 2009.8.Vissers LE., Veltman JA., van Kessel AG., Brunner HG. Identification of disease genes by whole genome CGH arrays. Hum Mol Genet. 2005. 14(Review Issue 2):R. 215–23.
Article9.Ullmann R., Turner G., Kirchhoff M., Chen W., Tonge B., Rosenberg C, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007. 28:674–82.
Article10.Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998. 14:417–22.
Article11.Inoue K., Lupski JR. Molecular mechanisms for genomic disorders. Annu Rev Genomics Hum Genet. 2002. 3:199–242.
Article12.McCarroll SA., Hadnott TN., Perry GH., Sabeti PC., Zody MC., Barrett JC, et al. Common deletion polymorphisms in the human genome. Nat Genet. 2006. 38:86–92.
Article13.Conrad DF., Andrews TD., Carter NP., Hurles ME., Pritchard JK. A high-resolution survey of deletion polymorphism in the human genome. Nat Genet. 2006. 38:75–81.
Article14.Allen KM., Gleeson JG., Bagrodia S., Partington MW., MacMillan JC., Cerione RA, et al. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998. 20:25–30.
Article15.Tarpey P., Parnau J., Blow M., Woffendin H., Bignell G., Cox C, et al. Mutations in the DLG3 gene cause nonsyndromic X-linked mental retardation. Am J Hum Genet. 2004. 75:318–24.
Article16.Monahan P., Rybak S., Raetzman LT. The notch target gene HES1 regulates cell cycle inhibitor expression in the developing pituitary. Endocrinology. 2009. 150:4386–94.17.Rochais F., Dandonneau M., Mesbah K., Jarry T., Mattei MG., Kelly RG. Hes1 is expressed in the second heart field and is required for outflow tract development. PLoS One. 2009. 4:e6267.
Article18.Dunn LL., Sekyere EO., Rahmanto YS., Richardson DR. The function of melanotransferrin: a role in melanoma cell proliferation and tumorigenesis. Carcinogenesis. 2006. 27:2157–69.
Article19.Sekyere EO., Dunn LL., Rahmanto YS., Richardson DR. Role of melanotransferrin in iron metabolism: studies using targeted gene disruption in vivo. Blood. 2006. 107:2599–601.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A de novo Proximal 6q Deletion Confirmed by Array Comparative Genomic Hybridization
- A Case of Chromosome 1q44 Deletion with Microcephaly and Multiple Congenital Anomalies
- Overgrowth Syndrome with 9q22.3 Microdeletion Detected by Microarray Comparative Genomic Hybridization
- Characteristics of Interstitial Deletion in Chromosome 4q Confirmed by Array Comparative Genomic Hybridization: A Case Report and Literature Review
- 14q32.33 Deletion Identified by array-CGH in a 5-year old-girl with Seizure
