Ann Lab Med.
2024 May;44(3):271-278. 10.3343/alm.2023.0152.
Re-evaluation of a Fibrillin-1 Gene Variant of Uncertain Significance Using the ClinGen Guidelines
- Affiliations
-
- 1Department of Laboratory Medicine, Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
- 2Department of Laboratory Medicine, Gangnam Severance Hospital, Yonsei University College of Medicine, Seoul, Korea
- KMID: 2555698
- DOI: http://doi.org/10.3343/alm.2023.0152
Abstract
- Background
Marfan syndrome (MFS) is caused by fibrillin-1 gene (FBN1) variants. Mutational hotspots and/or well-established critical functional domains of FBN1 include cysteine residues, calcium-binding consensus sequences, and amino acids related to interdomain packaging. Previous guidelines for variant interpretation do not reflect the features of genes or related diseases. Using the Clinical Genome Resource (ClinGen) FBN1 variant curation expert panel (VCEP), we re-evaluated FBN1 germline variants reported as variants of uncertain significance (VUSs).
Methods
We re-evaluated 26 VUSs in FBN1 reported in 161 patients with MFS. We checked the variants in the Human Genome Mutation Database, ClinVar, and VarSome databases and assessed their allele frequencies using the gnomAD database. Patients’ clinical information was reviewed.
Results
Four missense variants affecting cysteines (c.460T > C, c.1006T > C, c.5330G > C, and c.8020T > C) were reclassified as likely pathogenic and were assigned PM1_strong or PM1. Two intronic variants were reclassified as benign by granting BA1 (stand-alone). Four missense variants were reclassified as likely benign. BP5 criteria were applied in cases with an alternate molecular basis for disease, one of which (c.7231G > A) was discovered alongside a pathogenic de novo COL3A1 variant (c.1988G > T, p.Gly633Val).
Conclusions
Considering the high penetrance of FBN1 variants and clinical variability of MFS, the detection of pathogenic variants is important. The ClinGen FBN1 VCEP encompasses mutational hotspots and/or well-established critical functional domains and adjusts the criteria specifically for MFS; therefore, it is beneficial not only for identifying pathogenic FBN1 variants but also for distinguishing these variants from those that cause other connective tissue disorders with overlapping clinical features.
Figure
-
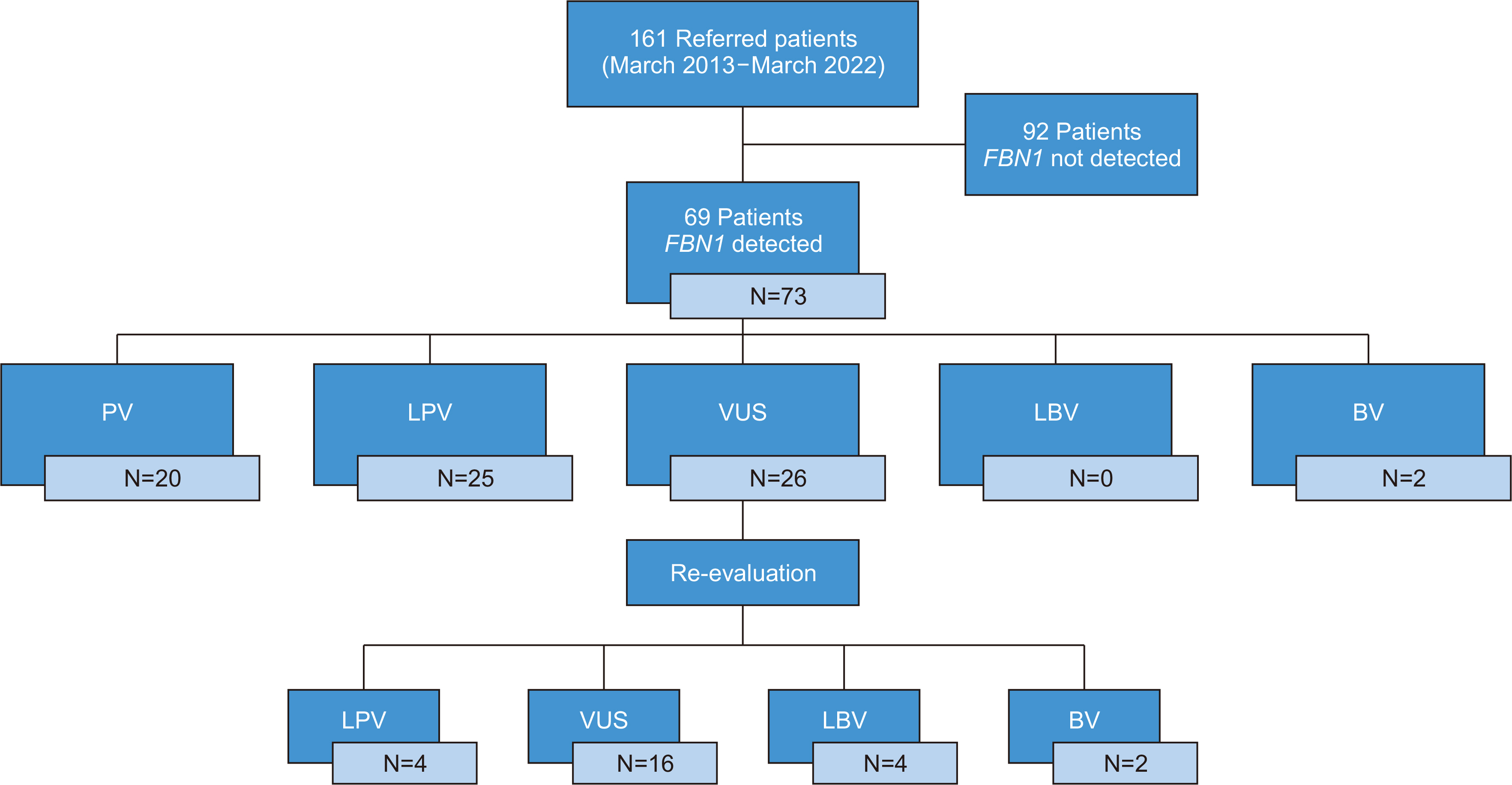
Fig. 1 Classification of reported FBN1 variants according to the ACMG/AMP guidelines. In this study, 161 patients diagnosed as having MFS, suspected of having MFS (presenting marfanoid features), or with aortic aneurysm/dissection were enrolled. Seventy-three FBN1 variants were identified in 69 of these patients. The ACMG/AMP guidelines were used to classify the 73 variants as PV, LPV, VUS, LBV, or BV. Among 26 VUSs, 24 different types were reported in 26 patients. Abbreviations: ACMG/AMP, American College of Medical Genetics and Association for Molecular Pathology; MFS, marfan syndrome; PV, pathogenic variant; LPV, likely pathogenic variant; VUS, variant of uncertain significance; LBV, likely benign variant; BV, benign variant.
Reference
-
References
1. Dean JC. 2007; Marfan syndrome: clinical diagnosis and management. Eur J Hum Genet. 15:724–33. DOI: 10.1038/sj.ejhg.5201851. PMID: 17487218.2. Sakai LY, Keene DR, Engvall E. 1986; Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 103:2499–509. DOI: 10.1083/jcb.103.6.2499. PMID: 3536967. PMCID: PMC2114568.3. Schrenk S, Cenzi C, Bertalot T, Conconi MT, Di Liddo R. 2018; Structural and functional failure of fibrillin-1 in human diseases (Review). Int J Mol Med. 41:1213–23. DOI: 10.3892/ijmm.2017.3343. PMID: 29286095.4. Vollbrandt T, Tiedemann K, El-Hallous E, Lin G, Brinckmann J, John H, et al. 2004; Consequences of cysteine mutations in calcium-binding epidermal growth factor modules of fibrillin-1. J Biol Chem. 279:32924–31. DOI: 10.1074/jbc.M405239200. PMID: 15161917.5. Zhang M, Chen Z, Chen T, Sun X, Jiang Y. 2021; Cysteine substitution and calcium-binding mutations in FBN1 cbEGF-like domains are associated with severe ocular involvement in patients with congenital ectopia lentis. Front Cell Dev Biol. 9:816397. DOI: 10.3389/fcell.2021.816397. PMID: 35237611. PMCID: PMC8882981.6. Baudhuin LM, Kluge ML, Kotzer KE, Lagerstedt SA. 2019; Variability in gene-based knowledge impacts variant classification: an analysis of FBN1 missense variants in ClinVar. Eur J Hum Genet. 27:1550–60. DOI: 10.1038/s41431-019-0440-3. PMID: 31227806. PMCID: PMC6777626.7. Rivera-Muñoz EA, Milko LV, Harrison SM, Azzariti DR, Kurtz CL, Lee K, et al. 2018; ClinGen Variant Curation Expert Panel experiences and standardized processes for disease and gene-level specification of the ACMG/AMP guidelines for sequence variant interpretation. Hum Mutat. 39:1614–22. DOI: 10.1002/humu.23645. PMID: 30311389. PMCID: PMC6225902.8. De Backer J. ClinGen FBN1 Expert Panel Specifications to the ACMG/AMP Variant Interpretation Guidelines Version 1. https://clinicalgenome.org/docs/application-for-variant-curation-expert-panel-status. Updated on Feb 2022.9. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. 2016; REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 99:877–85. DOI: 10.1016/j.ajhg.2016.08.016. PMID: 27666373. PMCID: PMC5065685.10. Chen ZR, Bao MH, Wang XY, Yang YM, Huang B, Han ZL, et al. 2021; Genetic variants in Chinese patients with sporadic Stanford type A aortic dissection. J Thorac Dis. 13:4008–22. DOI: 10.21037/jtd-20-2758. PMID: 34422331. PMCID: PMC8339749.11. Takeda N, Inuzuka R, Maemura S, Morita H, Nawata K, Fujita D, et al. 2018; Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ Genom Precis Med. 11:e002058. DOI: 10.1161/CIRCGEN.118.002321.12. Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, et al. 2007; Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 81:454–66. DOI: 10.1086/520125. PMID: 17701892. PMCID: PMC1950837.13. Pisano C, Balistreri CR, Nardi P, Altieri C, Bertoldo F, Buioni D, et al. 2020; Risk of aortic dissection in patients with ascending aorta aneurysm: a new biological, morphological, and biomechanical network behind the aortic diameter. Vessel Plus. 4:33. DOI: 10.20517/2574-1209.2020.21.14. Biggin A, Holman K, Brett M, Bennetts B, Adès L. 2004; Detection of thirty novel FBN1 mutations in patients with Marfan syndrome or a related fibrillinopathy. Hum Mutat. 23:99. DOI: 10.1002/humu.9207. PMID: 14695540.15. Comeglio P, Johnson P, Arno G, Brice G, Evans A, Aragon-Martin J, et al. 2007; The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat. 28:928. DOI: 10.1002/humu.9505. PMID: 17657824.16. Jalkh N, Mehawej C, Chouery E. 2020; Actionable exomic secondary findings in 280 Lebanese participants. Front Genet. 11:208. DOI: 10.3389/fgene.2020.00208. PMID: 32231684. PMCID: PMC7083077.17. Regalado ES, Guo DC, Prakash S, Bensend TA, Flynn K, Estrera A, et al. 2015; Aortic disease presentation and outcome associated with ACTA2 mutations. Circ Cardiovasc Genet. 8:457–64. DOI: 10.1161/CIRCGENETICS.114.000943. PMID: 25759435. PMCID: PMC4601641.18. Diness BR, Palmquist RN, Norling R, Hove H, Bundgaard H, Hertz JM, et al. 2020; Expanding the cerebrovascular phenotype of the p.R258H variant in ACTA2 related hereditary thoracic aortic disease (HTAD). J Neurol Sci. 415:116897. DOI: 10.1016/j.jns.2020.116897. PMID: 32464348.19. Kathiravel U, Keyser B, Hoffjan S, Kötting J, Müller M, Sivalingam S, et al. 2013; High-density oligonucleotide-based resequencing assay for mutations causing syndromic and non-syndromic forms of thoracic aortic aneurysms and dissections. Mol Cell Probes. 27:103–8. DOI: 10.1016/j.mcp.2012.10.002. PMID: 23142374.20. Yang H, Luo M, Fu Y, Cao Y, Yin K, Li W, et al. 2016; Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep. 6:33002. DOI: 10.1038/srep33002. PMID: 27611364. PMCID: PMC5017237.21. Milewicz DM, Østergaard JR, Ala-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. 2010; 152A:2437–43. DOI: 10.1002/ajmg.a.33657. PMID: 20734336. PMCID: PMC3573757.22. Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. 2009; Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 84:617–27. DOI: 10.1016/j.ajhg.2009.04.007. PMID: 19409525. PMCID: PMC2680995.23. Groth KA, Von Kodolitsch Y, Kutsche K, Gaustadnes M, Thorsen K, Andersen NH, et al. 2017; Evaluating the quality of Marfan genotype-phenotype correlations in existing FBN1 databases. Genet Med. 19:772–7. DOI: 10.1038/gim.2016.181. PMID: 27906200.24. Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, et al. 2017; The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 175:8–26. DOI: 10.1002/ajmg.c.31552. PMID: 28306229.25. Yen JL, Lin SP, Chen MR, Niu DM. 2006; Clinical features of Ehlers-Danlos syndrome. J Formos Med Assoc. 105:475–80. DOI: 10.1016/S0929-6646(09)60187-X. PMID: 16801035.26. Clapp IM, Paul KM, Beck EC, Nho SJ. 2021; Hypermobile disorders and their effects on the hip joint. Front Surg. 8:596971. DOI: 10.3389/fsurg.2021.596971. PMID: 33842528. PMCID: PMC8027473.27. Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. 2014; Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 16:881–8. DOI: 10.1038/gim.2014.72. PMID: 24922459.28. Pepin MG, Murray ML, Bailey S, Leistritz-Kessler D, Schwarze U, Byers PH. 2016; The challenge of comprehensive and consistent sequence variant interpretation between clinical laboratories. Genet Med. 18:20–4. DOI: 10.1038/gim.2015.31. PMID: 25834947.29. Najafi A, Caspar SM, Meienberg J, Rohrbach M, Steinmann B, Matyas G. 2020; Variant filtering, digenic variants, and other challenges in clinical sequencing: a lesson from fibrillinopathies. Clin Genet. 97:235–45. DOI: 10.1111/cge.13640. PMID: 31506931. PMCID: PMC7004123.30. Hayward C, Porteous ME, Brock DJ. 1997; Mutation screening of all 65 exons of the fibrillin-1 gene in 60 patients with Marfan syndrome: report of 12 novel mutations. Hum Mutat. 10:280–9. DOI: 10.1002/(SICI)1098-1004(1997)10:4<280::AID-HUMU3>3.0.CO;2-L.31. Yang RQ, Jabbari J, Cheng XS, Jabbari R, Nielsen JB, Risgaard B, et al. 2014; New population-based exome data question the pathogenicity of some genetic variants previously associated with Marfan syndrome. BMC Genet. 15:74. DOI: 10.1186/1471-2156-15-74. PMID: 24941995. PMCID: PMC4070351.32. Groth KA, Gaustadnes M, Thorsen K, Østergaard JR, Jensen UB, Gravholt CH, et al. 2016; Difficulties in diagnosing Marfan syndrome using current FBN1 databases. Genet Med. 18:98–102. DOI: 10.1038/gim.2015.32. PMID: 25812041.33. Overwater E, Marsili L, Baars MJH, Baas AF, van de Beek I, Dulfer E, et al. 2018; Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum Mutat. 39:1173–92. DOI: 10.1002/humu.23565. PMID: 29907982. PMCID: PMC6175145.34. Na R, Hong J, Gu H, Lee W, Lee JL, Chun S, et al. 2022; RNA Sequencing Provides Evidence for Pathogenicity of a Novel CHEK2 Splice Variant (C.1009-7T>G). Ann Lab Med. 42:380–3. DOI: 10.3343/alm.2022.42.3.380. PMID: 34907112. PMCID: PMC8677473.35. Park SY, Lee JM, Kim MJ, Chung NG, Lee JB, Kim Y, et al. 2023; Validation of Pathogenicity of Gene Variants in Fanconi Anemia Using Patient-derived Dermal Fibroblasts. Ann Lab Med. 43:127–31. DOI: 10.3343/alm.2023.43.1.127. PMID: 36045072. PMCID: PMC9467830.36. Gu H, Hong J, Lee W, Kim SB, Chun S, Min WK. 2022; RNA Sequencing for Elucidating an Intronic Variant of Uncertain Significance (SDHD c.314+ 3A>T) in Splicing Site Consensus Sequences. Ann Lab Med. 42:376–9. DOI: 10.3343/alm.2022.42.3.376. PMID: 34907111. PMCID: PMC8677484.37. Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL. 2017; Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg. 6:582–94. DOI: 10.21037/acs.2017.11.03. PMID: 29270370. PMCID: PMC5721110.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Re-evaluation of the LDLR Gene Variants of Uncertain Significance Using ClinGen Guideline
- Is There a Relationship Between Pelvic Organ Prolapse and Tissue Fibrillin-1 Levels?
- Importance of family segregation in the American College of Medical Genetics and Genomics and Association of Molecular Pathology guidelines: Case of a Korean family with autosomal dominant polycystic disease
- Applying Functional Assay Evidence to Interpret Sequence Variants Identified in Hereditary Cancer Genes
- Challenges and Considerations in Sequence Variant Interpretation for Mendelian Disorders
