First Case of Peroxisomal D-bifunctional Protein Deficiency with Novel HSD17B4 Mutations and Progressive Neuropathy in Korea
- Affiliations
-
- 1Department of Pediatrics, Chungnam National University College of Medicine, Chungnam National University Hospital, Daejeon, Korea
- 2Department of Pediatrics, College of Medicine, Hallym University, Kangdong Sacred Heart Hospital, Seoul, Korea
- 3Department of Emergency Medicine, Seoul National University Hospital, Seoul, Korea
- 4Department of Pediatrics, Daejeon Eulji Medical Center, Eulji University, Daejeon, Korea
- 5Department of Pediatrics, Chungnam National University College of Medicine, Chungnam National University Sejong Hospital, Sejong, Korea
- KMID: 2507612
- DOI: http://doi.org/10.3346/jkms.2020.35.e357
Abstract
- Peroxisomal D-bifunctional protein (DBP), encoded by the HSD17B4 gene, catalyzes β-oxidation of very long chain fatty acids (VLCFAs). The deficiency of this peroxisomal enzyme leads to the accumulation of VLCFAs, causing multisystemic manifestations including the brain, retina, adrenal gland, hearing, and skeletal system. Herein, we report the first Korean neonatal case of peroxisomal DBP deficiency and the clinical prognosis over 2 years. This patient showed craniofacial dysmorphism, club foot, and seizures with cyanosis one day after birth. Elevated VLCFAs levels were indicative of a peroxisomal disorder. Targeted exome sequencing was performed and two missense mutations p.Asp117Val and p.Phe279Ser in the HSD17B4 gene were identified. The patient had type III DBP deficiency; therefore, docosahexaenoic acid and non-soluble vitamins were administered. However, progressive nystagmus, optic nerve atrophy, and bilateral hearing defects were observed and follow-up brain imaging revealed leukodystrophy and brain atrophy. Multiple anti-epileptic drugs were required to control the seizures. Over two years, the patient achieved normal growth with home ventilation and tube feeding. Hereby, the subject's parents had support during the second pregnancy from the proven molecular information. Moreover, targeted exome sequencing is an effective diagnostic approach, considering genetic heterogeneity of Zellweger spectrum disorders.
Keyword
Figure
-
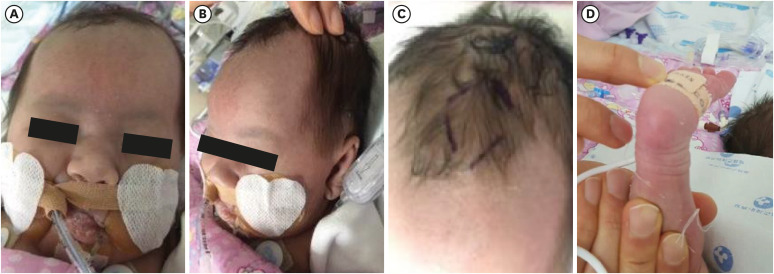
Fig. 1 Clinical photographs of patient. Patient showed (A) typical box-shaped face, (B) large fontanelle, (C) midfacial hypoplasia, and (D) club foot. The figures are published with agreement of parents.
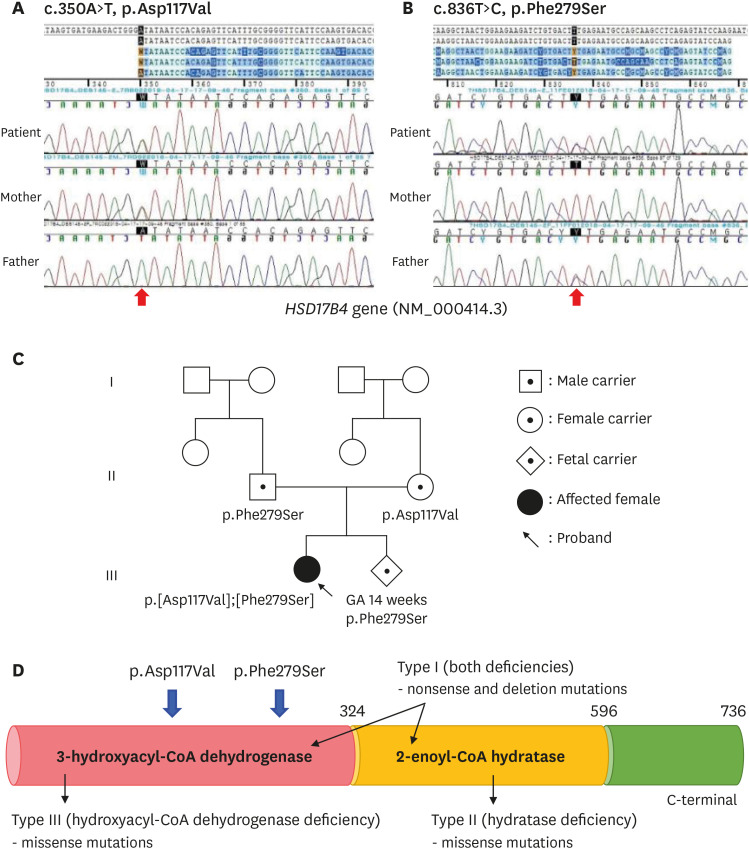
Fig. 2 Partial genomic DNA sequences of HSD17B4 (NM_000414.3) of patients and parents. The result showed compound heterozygote mutation of (A) c.350A>T (p.Asp117Val) and (B) c.836T>C (p.Phe279Ser), and each mutation was inherited from subject parents. (C) Pedigree of patients with D-bifunctional protein deficiency and (D) the locus of two variants representative of 3-hydroxyacyl-CoA dehydrogenase deficiency.
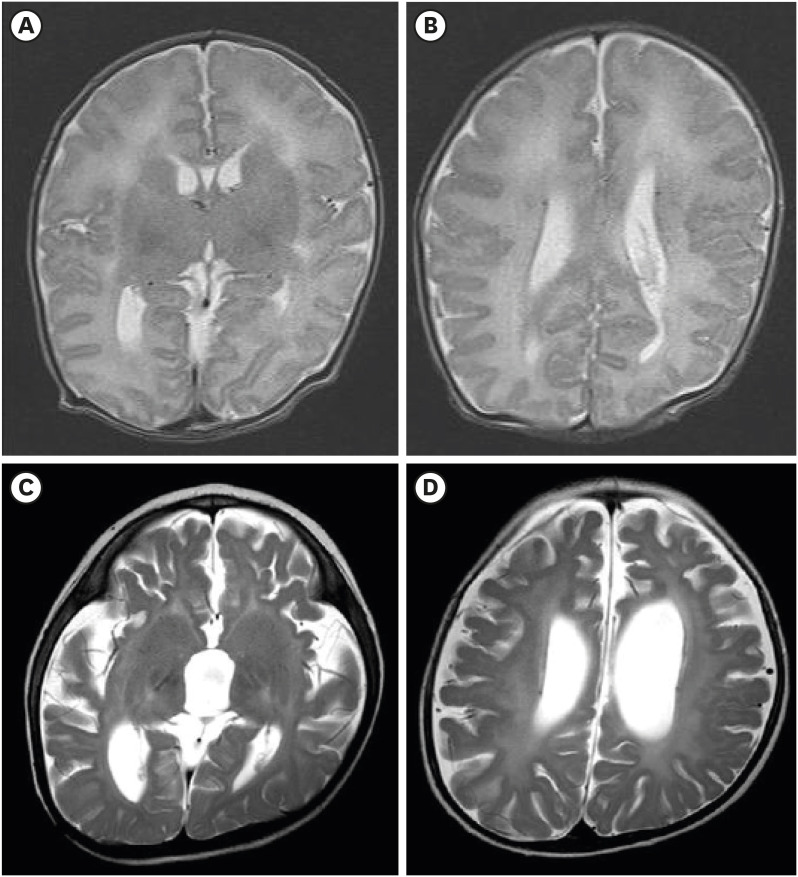
Fig. 3 Brain MRI of the patient. (A, B) Axial T2-image showing bilateral frontal and sylvian polymicrogyria. (C, D) Follow-up images of brain MRI of the patient showing diffuse increased T2 signal intensity and the atrophy of the brain. The patient's parents provided written informed consent for publication of the patient and family information.MRI = magnetic resonance imaging.
Reference
-
1. Ferdinandusse S, Denis S, Mooyer PA, Dekker C, Duran M, Soorani-Lunsing RJ, et al. Clinical and biochemical spectrum of D-bifunctional protein deficiency. Ann Neurol. 2006; 59(1):92–104. PMID: 16278854.
Article2. Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, et al. Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis. Am J Hum Genet. 2006; 78(1):112–124. PMID: 16385454.
Article3. Wanders RJ. Peroxisomal disorders: Improved laboratory diagnosis, new defects and the complicated route to treatment. Mol Cell Probes. 2018; 40:60–69. PMID: 29438773.
Article4. Amor DJ, Marsh AP, Storey E, Tankard R, Gillies G, Delatycki MB, et al. Heterozygous mutations in HSD17B4 cause juvenile peroxisomal D-bifunctional protein deficiency. Neurol Genet. 2016; 2(6):e114. PMID: 27790638.5. Watkins PA, Chen WW, Harris CJ, Hoefler G, Hoefler S, Blake DC Jr, et al. Peroxisomal bifunctional enzyme deficiency. J Clin Invest. 1989; 83(3):771–777. PMID: 2921319.
Article6. Nascimento J, Mota C, Lacerda L, Pacheco S, Chorão R, Martins E, et al. D-bifunctional protein deficiency: a cause of neonatal onset seizures and hypotonia. Pediatr Neurol. 2015; 52(5):539–543. PMID: 25882080.
Article7. Konkoľová J, Petrovič R, Chandoga J, Repiský M, Zelinková H, Kršiaková J, et al. Peroxisomal D-bifunctional protein deficiency: first case reports from Slovakia. Gene. 2015; 568(1):61–68. PMID: 25967389.
Article8. Ghirri P, Vuerich M, Ferdinandusse S, Waterham HR, Guzzetta A, Bianchi MC, et al. A case of D-bifunctional protein deficiency: clinical, biochemical and molecular investigations. Pediatr Int. 2011; 53(4):583–587. PMID: 21851493.
Article9. Incecik F, Mungan NO. D-bifunctional protein deficiency: a case report of a Turkish child. Ann Indian Acad Neurol. 2019; 22(1):119–121. PMID: 30692775.
Article10. Khromykh A, Solomon BD, Bodian DL, Leon EL, Iyer RK, Baker RL, et al. Diagnosis of D-bifunctional protein deficiency through whole-genome sequencing: implications for cost-effective care. Mol Syndromol. 2015; 6(3):141–146. PMID: 26733776.11. McMillan HJ, Worthylake T, Schwartzentruber J, Gottlieb CC, Lawrence SE, Mackenzie A, et al. Specific combination of compound heterozygous mutations in 17β-hydroxysteroid dehydrogenase type 4 (HSD17B4) defines a new subtype of D-bifunctional protein deficiency. Orphanet J Rare Dis. 2012; 7(1):90. PMID: 23181892.12. Farkas A, Al-Ramadhani R, McDonald K, Jordan M, Joyner D. Unusual Clinical course and imaging of D-bifunctional protein deficiency, a rare leukodystrophy. Pediatr Neurol. 2019; 90:70–71. PMID: 30396834.
Article13. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010; 86(5):749–764. PMID: 20466091.
Article14. Vasudevan P, Suri M. A clinical approach to developmental delay and intellectual disability. Clin Med (Lond). 2017; 17(6):558–561. PMID: 29196358.
Article15. van Karnebeek CD, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. 2012; 105(3):368–381. PMID: 22212131.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Peroxisomal disorders
- A Case of Wernicke's Encephalopathy Presenting as Acute Bilateral Wrist Drop
- A Case of Central Retinal Vein Occlusion by Protein C Deficiency
- Childhood Ischemic Stroke Associated with Protein S Deficiency: A case report
- Subacute Combined Degeneration with Motor Neuropathy Following Nitrous Oxide Abuse
