Acquired Resistance of MET-Amplified Non-small Cell Lung Cancer Cells to the MET Inhibitor Capmatinib
- Affiliations
-
- 1Cancer Research Institute, Seoul National University College of Medicine, Seoul, Korea. kimdw@snu.ac.kr
- 2Department of Internal Medicine, Seoul National University Hospital, Seoul, Korea.
- KMID: 2454287
- DOI: http://doi.org/10.4143/crt.2018.052
Abstract
- PURPOSE
Amplified mesenchymal-epithelial transition factor, MET, is a receptor tyrosine kinase (RTK) that has been considered a druggable target in non-small cell lung cancer (NSCLC). Although multiple MET tyrosine kinase inhibitors (TKIs) are being actively developed for MET-driven NSCLC, the mechanisms of acquired resistance to MET-TKIs have not been well elucidated. To understand the mechanisms of resistance and establish therapeutic strategies, we developed an in vitro model using the MET-amplified NSCLC cell line EBC-1.
MATERIALS AND METHODS
We established capmatinib-resistant NSCLC cell lines and identified alternative signaling pathways using 3"² mRNA sequencing and human phospho-RTK arrays. Copy number alterations were evaluated by quantitative polymerase chain reaction and cell proliferation assay; activation of RTKs and downstream effectors were compared between the parental cell line EBC-1 and the resistant cell lines.
RESULTS
We found that EBC-CR1 showed an epidermal growth factor receptor (EGFR)"’dependent growth and sensitivity to afatinib, an irreversible EGFR TKI. EBC-CR2 cells that had overexpression of EGFR-MET heterodimer dramatically responded to combined capmatinib with afatinib. In addition, EBC-CR3 cells derived from EBC-CR1 cells that activated EGFR with amplified phosphoinositide-3 kinase catalytic subunit α (PIK3CA) were sensitive to combined afatinib with BYL719, a phosphoinositide 3-kinase α (PI3Kα) inhibitor.
CONCLUSION
Our in vitro studies suggested that activation of EGFR signaling and/or genetic alteration of downstream effectors like PIK3CA were alternative resistance mechanisms used by capmatinib-resistant NSCLC cell lines. In addition, combined treatments with MET, EGFR, and PI3Kα inhibitors may be effective therapeutic strategies in capmatinib-resistant NSCLC patients.
Keyword
MeSH Terms
-
Carcinoma, Non-Small-Cell Lung*
Catalytic Domain
Cell Line
Cell Proliferation
Humans
In Vitro Techniques
Parents
Phosphotransferases
Polymerase Chain Reaction
Protein-Tyrosine Kinases
Receptor, Epidermal Growth Factor
RNA, Messenger
Phosphotransferases
Protein-Tyrosine Kinases
RNA, Messenger
Receptor, Epidermal Growth Factor
Figure
-
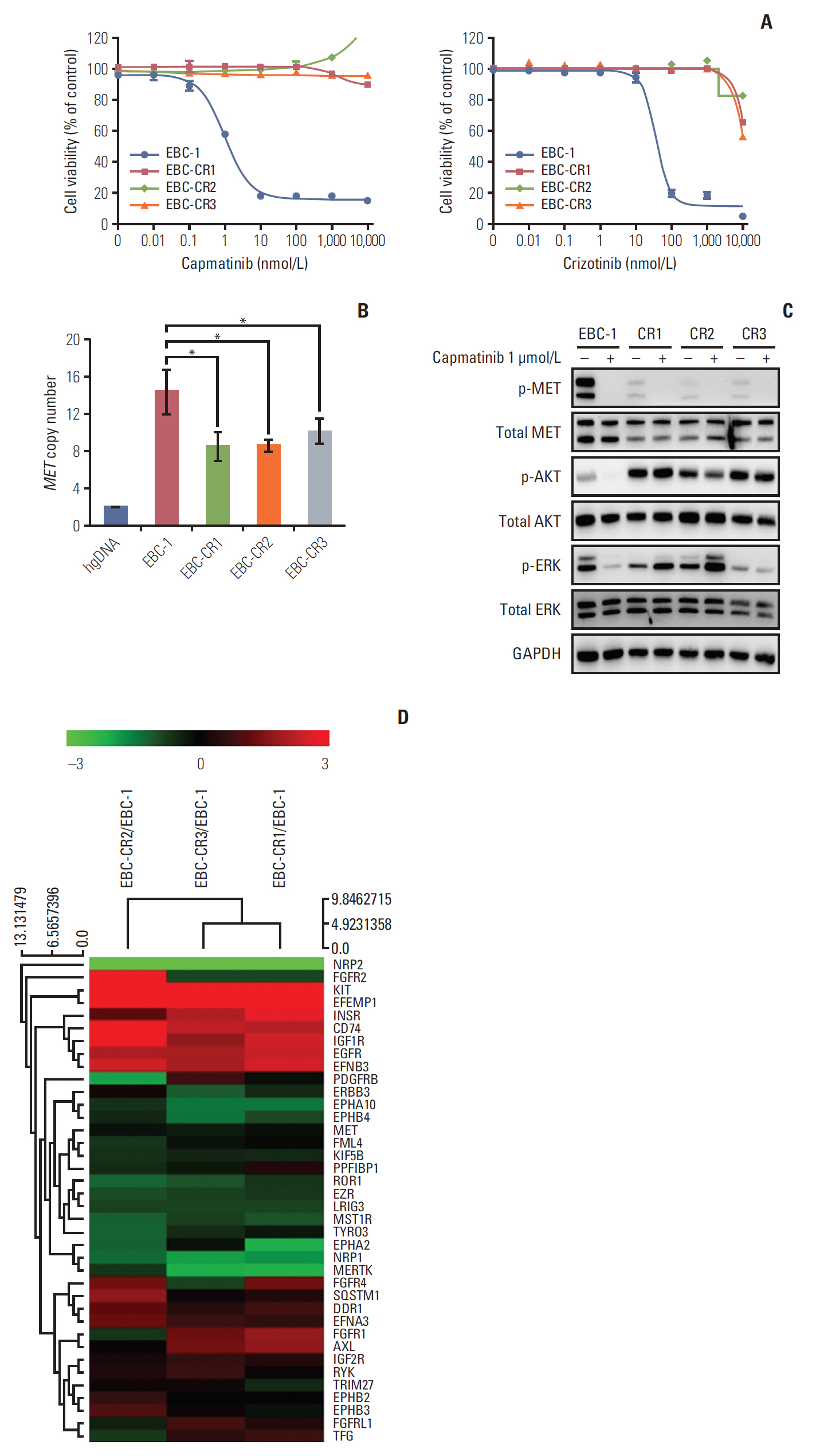
Fig. 1. Molecular characterization of capmatinib-resistant cell lines. (A) Capmatinib-resistant cell lines (EBC-CR1, EBC-CR2, and EBC-CR3) were derived from EBC-1, which is a non-small cell lung cancer cell line that harbors mesenchymal-epithelial transition factor (MET) amplification. Cell lines were treated with capmatinib and crizotinib for 72 hours and growth inhibition was determined by cell viability assay (Ez-cytox). Tests were performed as three independent experiments. The EBC-CR1, -CR2, and -CR3 cell lines showed resistance to capmatinib and cross-resistance to crizotinib. (B) The resistant cell lines showed significant MET copy number loss after long-term treatment with capmatinib (*p < 0.05). MET copy number was confirmed by quantitative polymerase chain reaction. hgDNA, human genomic DNA (C) Capmatinib-resistant cells had persistent expression of phosphorylated ERK1/2 and AKT in the presence of capmatinib. EBC-1 and the resistant cell lines were treated with capmatinib at 1 μmol/L for 2 hours then analyzed by Western blot. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as the loading control for the Western blot. (D) Hierarchical clustering analysis of the resistant cell lines revealed a cluster containing seven receptor tyrosine kinases (RTKs) that was increased in all resistant cell lines. The values for clustering analysis fulfilled the requirements of normalized read counts (log2) > 6 and p < 0.05. The fold changes in expression of these RTKs are indicated in Table 1. Each color represents relative gene expression with the highest expression as red, lowest expression as green, and median expression as black.
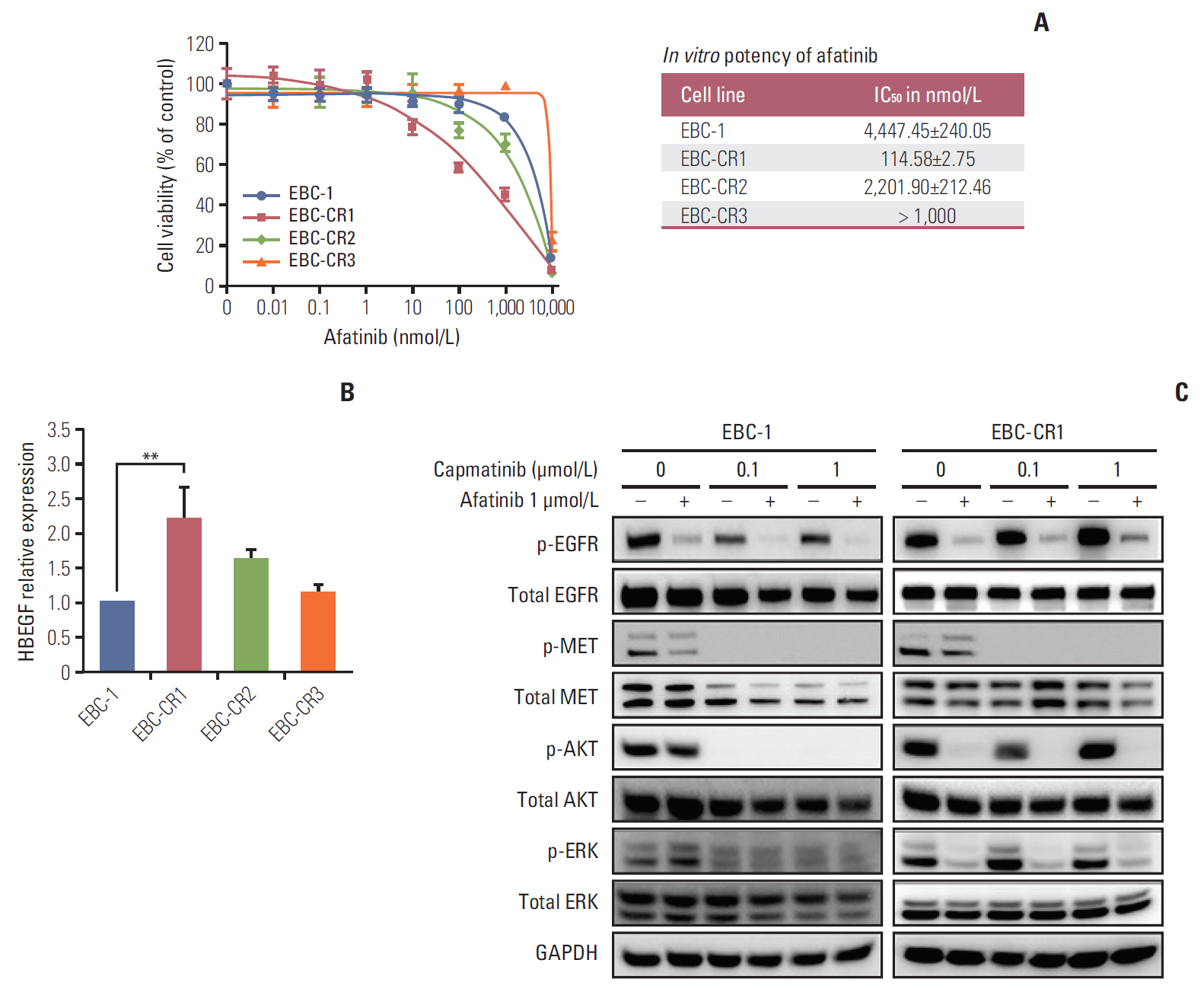
Fig. 2. Shift from mesenchymal-epithelial transition factor (MET) to epidermal growth factor receptor (EGFR) kinase pathway in EBC-CR1. (A) EBC-1 and resistant cell lines were treated with afatinib for 72 hours. The 50% inhibitory concentrations (IC50) were calculated using Sigma Plot 12.0; results are indicated as mean±standard deviation. (B) Heparin-binding epidermal growth factor‒like growth factor (HBEGF) expression was measured by quantitative reverse transcription polymerase chain reaction in three independent experiments. HBEGF expression was increased in the EBC-CR1 cell line compared to the parental and other capmatinib-resistant cell lines (**p < 0.01). (C) For Western blot, EBC-1 and EBC-CR1 cells were treated with serial 10-fold dilutions ranging from 0.1 to 1 μmol/L of capmatinib with or without afatinib at 1 μmol/L for 24 hours. EBC-CR1 showed almost complete dependency on downstream signaling due to afatinib treatment with or without capmatinib. Phosphorylation of AKT and ERK1/2 was efficiently inhibited by capmatinib in EBC-1 cells. GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
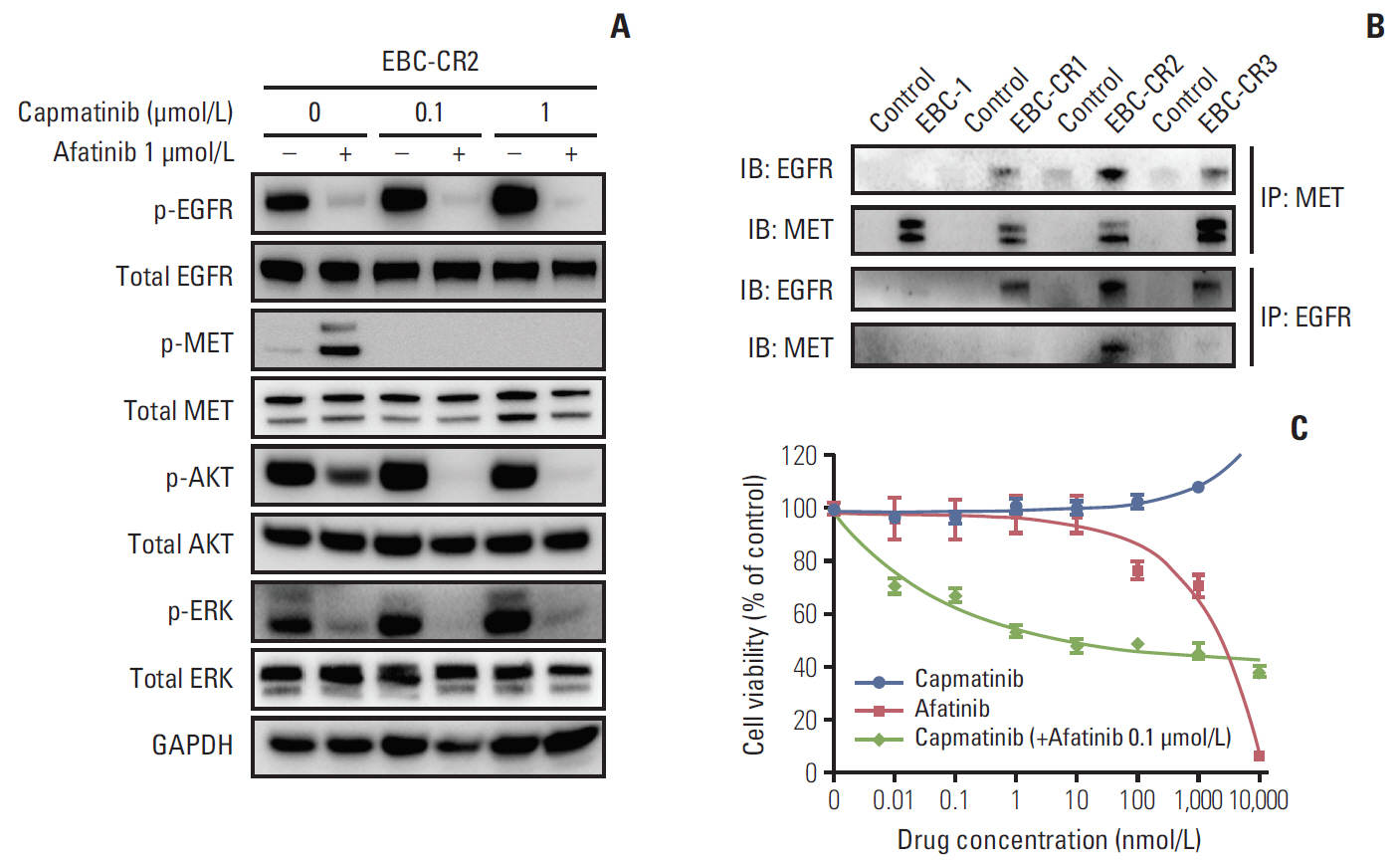
Fig. 3. Combined treatment with capmatinib and afatinib effectively inhibited phosphorylation of downstream signaling and proliferation in EBC-CR2 cells. (A) For Western blot, EBC-CR2 cells were treated with increasing concentrations of capmatinib with or without afatinib at 1 μmol/L for 24 hours. Capmatinib significantly increased epidermal growth factor receptor (EGFR) phosphorylation in a dose-dependent manner. The combination of capmatinib with afatinib at 1 μmol/L effectively inhibited phosphorylation of AKT and ERK at a low concentration of capmatinib, 100 nmol/L. (B) The heterodimerization of receptor tyrosine kinases was analyzed by co-immunoprecipitation. Although immunoprecipitation (IP) with mesenchymal-epithelial transition factor (MET) showed EGFR-MET interactions in EBC-CR1, 2, and 3 cells, IP with EGFR showed an interaction between EGFR and MET only in EBC-CR2 cells. Therefore, EBC-CR2 cells had more dominant EGFR-MET heterodimers than the other resistant cell lines. (C) EBC-CR2 cells treated with capmatinib, afatinib, and capmatinib with afatinib at 0.1 μmol/L, respectively, for 72 hours. Treatment with the combination of capmatinib with afatinib synergistically inhibited EBC-CR2 cell proliferation.
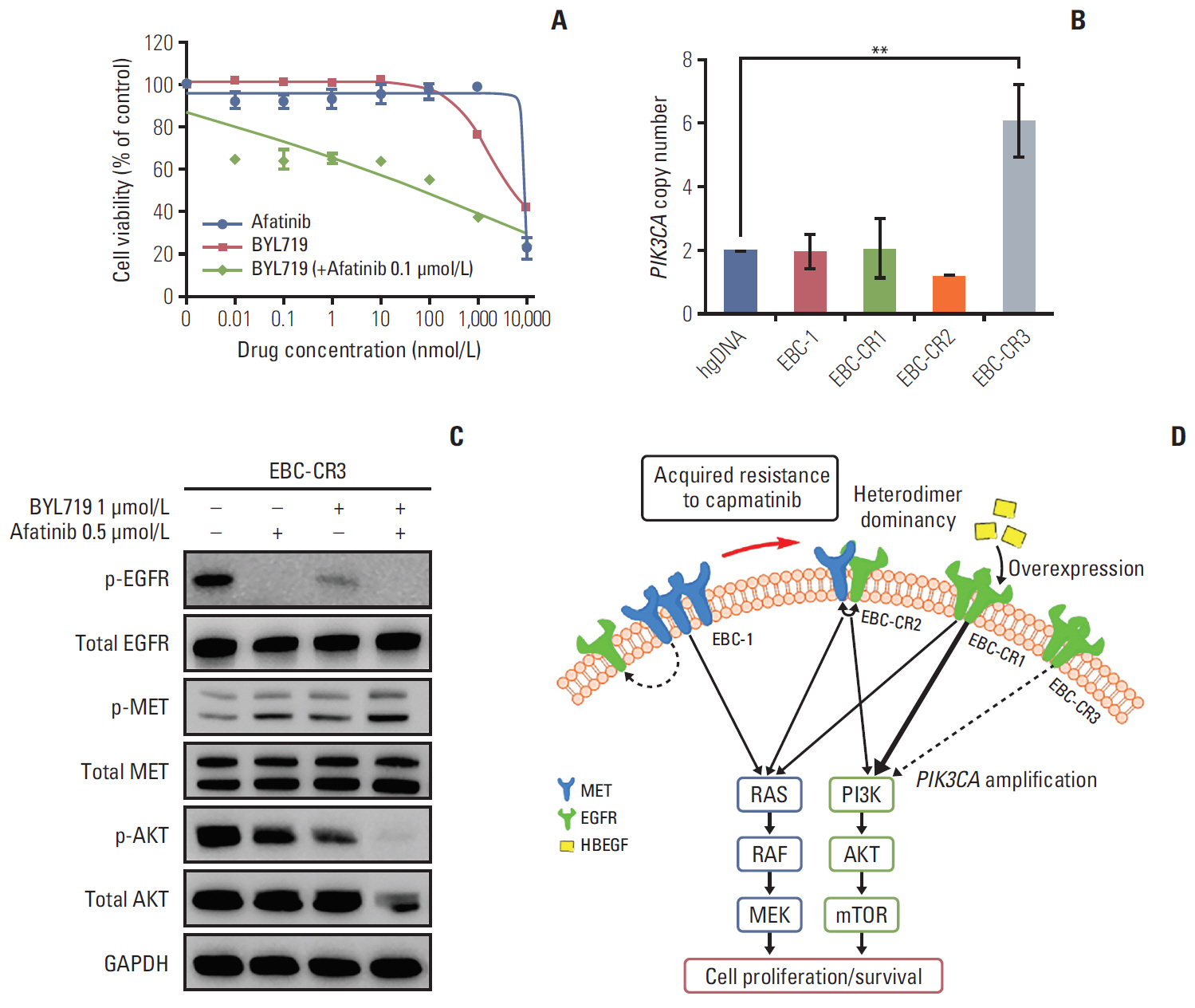
Fig. 4. PIK3CA amplification in EBC-CR3, resulting to afatinib-resistance and schematic model of resistant mechanisms to capmatinib in MET-amplified non-small cell lung cancer (NSCLC) cell lines. (A) Treatment of EBC-CR3 cells with afatinib, BYL719, and BYL719 with afatinib at 0.1 μmol/L, respectively, for 72 hours. BYL719 with afatinib synergistically inhibited proliferation of EBC-CR3 cells. (B) PIK3CA copy numbers in the cell lines were measured by quantitative polymerase chain reaction. Compared to the other cell lines and the human genomic DNA control, PIK3CA was amplified in the EBC-CR3 cell line (**p < 0.01). (C) Treatment of EBC-CR3 cells with afatinib 500 nmol/L, BYL719 1 μmol/L, or BYL719 with afatinib at 500 nmol/L, respectively, for 24 hours. The combination of BYL719 with afatinib completely inhibited AKT phosphorylation. (D) In a drug-sensitive cell line harboring MET amplification, cell proliferation and survival signals are highly dependent on constitutively activated MET kinase without ligand binding and/or homodimerization. In addition, MET activates the epidermal growth factor receptor (EGFR) signal pathway via multiple mechanisms. In contrast, the MET pathway cannot act as a survival signal and alternative pathways must be activated in capmatinib-resistant cells. We confirmed three different mechanisms of resistance to capmatinib in MET-dependent NSCLC cell lines. (1) heterodimerization between MET and EGFR, (2) increased EGFR and EGF-like growth factor (HBEGF) expression; and (3) PIK3CA amplification via activated EGFR-dependent PIK3CA stimulation, resulting in afatinib resistance. Each mechanism has different molecular characteristics and therapeutic strategies. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PI3K, phosphoinositide-3-kinase; mTOR, mammalian target of rapamycin.
Cited by 1 articles
-
MiR-1246 Promotes Metastasis and Invasion of A549 cells by Targeting GSK-3β‒Mediated Wnt/β-Catenin Pathway
Fan Yang, Hairong Xiong, Li Duan, Qian Li, Xin Li, Yongqin Zhou
Cancer Res Treat. 2019;51(4):1420-1429. doi: 10.4143/crt.2018.638.
Reference
-
References
1. Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004; 4:361–70.
Article2. Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008; 9:628–38.
Article3. Jo M, Stolz DB, Esplen JE, Dorko K, Michalopoulos GK, Strom SC. Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J Biol Chem. 2000; 275:8806–11.
Article4. Ma PC. MET receptor juxtamembrane exon 14 alternative spliced variant: novel cancer genomic predictive biomarker. Cancer Discov. 2015; 5:802–5.
Article5. Sadiq AA, Salgia R. MET as a possible target for non-small-cell lung cancer. J Clin Oncol. 2013; 31:1089–96.
Article6. Liu X, Wang Q, Yang G, Marando C, Koblish HK, Hall LM, et al. A novel kinase inhibitor, INCB28060, blocks c-MET-dependent signaling, neoplastic activities, and cross-talk with EGFR and HER-3. Clin Cancer Res. 2011; 17:7127–38.
Article7. Beau-Faller M, Ruppert AM, Voegeli AC, Neuville A, Meyer N, Guerin E, et al. MET gene copy number in non-small cell lung cancer: molecular analysis in a targeted tyrosine kinase inhibitor naive cohort. J Thorac Oncol. 2008; 3:331–9.8. Onozato R, Kosaka T, Kuwano H, Sekido Y, Yatabe Y, Mitsudomi T. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J Thorac Oncol. 2009; 4:5–11.
Article9. Onitsuka T, Uramoto H, Ono K, Takenoyama M, Hanagiri T, Oyama T, et al. Comprehensive molecular analyses of lung adenocarcinoma with regard to the epidermal growth factor receptor, K-ras, MET, and hepatocyte growth factor status. J Thorac Oncol. 2010; 5:591–6.
Article10. Park S, Koh J, Kim DW, Kim M, Keam B, Kim TM, et al. MET amplification, protein expression, and mutations in pulmonary adenocarcinoma. Lung Cancer. 2015; 90:381–7.
Article11. Comoglio PM, Giordano S, Trusolino L. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat Rev Drug Discov. 2008; 7:504–16.
Article12. Zou HY, Li Q, Lee JH, Arango ME, McDonnell SR, Yamazaki S, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007; 67:4408–17.
Article13. Brandes F, Schmidt K, Wagner C, Redekopf J, Schlitt HJ, Geissler EK, et al. Targeting cMET with INC280 impairs tumour growth and improves efficacy of gemcitabine in a pancreatic cancer model. BMC Cancer. 2015; 15:71.
Article14. Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015; 5:850–9.
Article15. Bang YJ, Su WC, Nam DH, Lim WT, Bauer TM, Brana I, et al. Phase I study of the safety and efficacy of INC280 in patients with advanced MET-dependent solid tumors. J Clin Oncol. 2014; 32(15 Suppl):2520.
Article16. Camaj P, Seeliger H, Ischenko I, Krebs S, Blum H, De Toni EN, et al. EFEMP1 binds the EGF receptor and activates MAPK and Akt pathways in pancreatic carcinoma cells. Biol Chem. 2009; 390:1293–302.
Article17. Yamamoto H, Shigematsu H, Nomura M, Lockwood WW, Sato M, Okumura N, et al. PIK3CA mutations and copy number gains in human lung cancers. Cancer Res. 2008; 68:6913–21.
Article18. Fritsch C, Huang A, Chatenay-Rivauday C, Schnell C, Reddy A, Liu M, et al. Characterization of the novel and specific PI3Kalpha inhibitor NVP-BYL719 and development of the patient stratification strategy for clinical trials. Mol Cancer Ther. 2014; 13:1117–29.19. Lutterbach B, Zeng Q, Davis LJ, Hatch H, Hang G, Kohl NE, et al. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 2007; 67:2081–8.
Article20. Lennerz JK, Kwak EL, Ackerman A, Michael M, Fox SB, Bergethon K, et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J Clin Oncol. 2011; 29:4803–10.
Article21. Egile C, Kenigsberg M, Delaisi C, Begassat F, Do-Vale V, Mestadier J, et al. The selective intravenous inhibitor of the MET tyrosine kinase SAR125844 inhibits tumor growth in MET-amplified cancer. Mol Cancer Ther. 2015; 14:384–94.
Article22. McDermott U, Pusapati RV, Christensen JG, Gray NS, Settleman J. Acquired resistance of non-small cell lung cancer cells to MET kinase inhibition is mediated by a switch to epidermal growth factor receptor dependency. Cancer Res. 2010; 70:1625–34.
Article23. Qi J, McTigue MA, Rogers A, Lifshits E, Christensen JG, Janne PA, et al. Multiple mutations and bypass mechanisms can contribute to development of acquired resistance to MET inhibitors. Cancer Res. 2011; 71:1081–91.
Article24. Benderra MA, Aspeslagh S, Postel-Vinay S, Bigot L, De Baere T, Loriot Y, et al. Acquired EGFR mutation as the potential resistance driver to crizotinib in a MET-mutated tumor. J Thorac Oncol. 2016; 11:e21–3.
Article25. Kwak EL, Ahronian LG, Siravegna G, Mussolin B, Borger DR, Godfrey JT, et al. Molecular heterogeneity and receptor coamplification drive resistance to targeted therapy in MET-amplified esophagogastric cancer. Cancer Discov. 2015; 5:1271–81.
Article26. Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003; 144:1032–44.
Article27. Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res. 2006; 66:10100–11.
Article28. Tanizaki J, Okamoto I, Sakai K, Nakagawa K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br J Cancer. 2011; 105:807–13.
Article29. Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017; 23:703–13.30. Spoerke JM, O'Brien C, Huw L, Koeppen H, Fridlyand J, Brachmann RK, et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res. 2012; 18:6771–83.
Article31. Liu X, Jia Y, Stoopler MB, Shen Y, Cheng H, Chen J, et al. Nextgeneration sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J Clin Oncol. 2016; 34:794–802.
Article32. Hellman A, Zlotorynski E, Scherer SW, Cheung J, Vincent JB, Smith DI, et al. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell. 2002; 1:89–97.
Article33. Piscitello D, Varshney D, Lilla S, Vizioli MG, Reid C, Gorbunova V, et al. AKT overactivation can suppress DNA repair via p70S6 kinase-dependent downregulation of MRE11. Oncogene. 2018; 37:427–38.
Article34. Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey HE, et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat Med. 2016; 22:262–9.
Article35. Chabon JJ, Simmons AD, Lovejoy AF, Esfahani MS, Newman AM, Haringsma HJ, et al. Circulating tumour DNA profiling reveals heterogeneity of EGFR inhibitor resistance mechanisms in lung cancer patients. Nat Commun. 2016; 7:11815.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- The Clinical Impact of Capmatinib in the Treatment of Advanced Non–Small Cell Lung Cancer with MET Exon 14 Skipping Mutation or Gene Amplification
- Inhibition of Ubiquitin-specific Peptidase 8 Suppresses Growth of Gefitinib-resistant Non-small Cell Lung Cancer Cells by Inducing Apoptosis
- Evaluation of MET alteration in EGFR-mutant non-small cell lung cancer patients treated with EGFR tyrosine kinase inhibitor from paired biopsy: A retrospective cohort study
- The Antitumor Effect of C-terminus of Hsp70-Interacting Protein via Degradation of c-Met in Small Cell Lung Cancer
- Mechanisms of Acquired Resistance to Epidermal Growth Factor Receptor Inhibitors and Overcoming Strategies in Lung Cancer
