Progression of GNE Myopathy Based on the Patient-Reported Outcome
- Affiliations
-
- 1Department of Neurology, Pusan National University Hospital, Busan, Korea.
- 2Department of Neurology, Pusan National University College of Medicine, Yangsan, Korea.
- 3Department of Neurology, Yonsei University College of Medicine, Seoul, Korea.
- 4Department of Neurology, Pusan National University Yangsan Hospital, Yangsan, Korea. shinzh@gmail.com
- KMID: 2451109
- DOI: http://doi.org/10.3988/jcn.2019.15.3.275
Abstract
- BACKGROUND AND PURPOSE
GNE myopathy is a rare progressive myopathy caused by biallelic mutations in the GNE gene, and frequently accompanied by rimmed vacuoles in muscle pathology. The initial symptom of foot drop or hip-girdle weakness eventually spreads to all limbs over a period of decades. Recent advances in pathophysiologic research have facilitated therapeutic trials aimed at resolving the core biochemical defect. However, there remains unsettled heterogeneity in its natural course, which confounds the analysis of therapeutic outcomes. We performed the first large-scale study of Korean patients with GNE myopathy.
METHODS
We gathered the genetic and clinical profiles of 44 Korean patients with genetically confirmed GNE myopathy. The clinical progression was estimated retrospectively based on a patient-reported questionnaire on the status of the functional joint sets and daily activities.
RESULTS
The wrist and neck were the last joints to lose antigravity functionality irrespective of whether the weakness started from the ankle or hip. Two-thirds of the patients could walk either independently or with an aid. The order of losing daily activities could be sorted from standing to eating. Patients with limb-girdle phenotype showed an earlier age at onset than those with foot-drop onset. Patients with biallelic kinase domain mutations tended to progress more rapidly than those with epimerase and kinase domain mutations.
CONCLUSIONS
The reported data can guide the clinical management of GNE myopathy, as well as provide perspective to help the development of clinical trials.
Keyword
MeSH Terms
Figure
-

Fig. 1 GNE mutations identified in our patients.

Fig. 2 Decrease in the number of functional joints. A: All of the functional joints in each of the patients. B: The functional joint sets were correlated with the disease duration (p<0.0001), and the mean yearly rate of decrease was −0.2581.
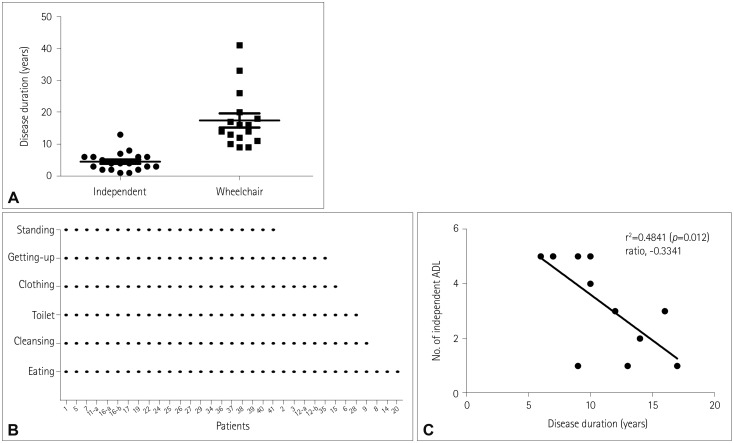
Fig. 3 Functional status during ambulation and daily activities. A: The disease duration clearly differed between the ambulatory and wheelchair-bound patients, at 4.5±0.7 and 17.4±2.2 years, respectively. B: All of the daily activities that could be performed independently by each of the patients. C: The number of independent daily activities was correlated with the disease duration (p=0.012). ADL: activities of daily living.
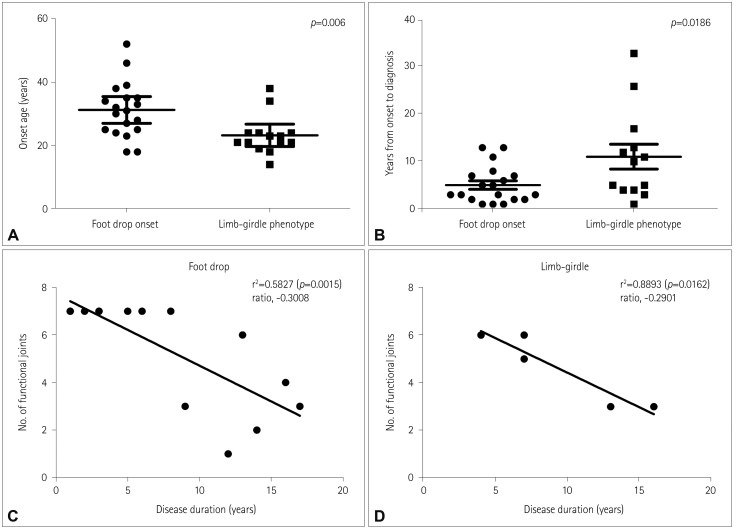
Fig. 4 Disease severity according to the types of initial symptoms. A: The onset age was significantly younger in the subgroup with limb-girdle phenotype. B: The duration from the onset to the diagnosis was significantly longer in the subgroup with limb-girdle phenotype. C and D: Disease progression as represented by the functional joint sets according to disease duration did not differ between the subgroups of foot-drop onset and limb-girdle phenotype (p=0.5731).

Fig. 5 Disease severity according to the domains of GNE mutations. A: The onset age did not differ significantly among the three subgroups of KD, ED/KD, and ED. B–D: Disease progression as represented by the number functional joint sets according to disease duration significantly rapid in the order of KD (B), ED/KD (C), and ED (D) (p=0.0062). ED: epimerase domain, KD: kinase domain.
Reference
-
1. Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001; 29:83–87. PMID: 11528398.
Article2. Huizing M, Carrillo-Carrasco N, Malicdan MC, Noguchi S, Gahl WA, Mitrani-Rosenbaum S, et al. GNE myopathy: new name and new mutation nomenclature. Neuromuscul Disord. 2014; 24:387–389. PMID: 24685570.
Article3. Park YE, Kim HS, Choi ES, Shin JH, Kim SY, Son EH, et al. Limbgirdle phenotype is frequent in patients with myopathy associated with GNE mutations. J Neurol Sci. 2012; 321:77–81. PMID: 22883483.
Article4. Nishino I, Carrillo-Carrasco N, Argov Z. GNE myopathy: current update and future therapy. J Neurol Neurosurg Psychiatry. 2015; 86:385–392. PMID: 25002140.
Article5. De Dios JK, Shrader JA, Joe GO, McClean JC, Williams K, Evers R, et al. Atypical presentation of GNE myopathy with asymmetric hand weakness. Neuromuscul Disord. 2014; 24:1063–1067. PMID: 25182749.
Article6. Celeste FV, Vilboux T, Ciccone C, De Dios JK, Malicdan MC, Leoyklang P, et al. Mutation update for GNE gene variants associated with GNE myopathy. Hum Mutat. 2014; 35:915–926. PMID: 24796702.7. Argov Z, Mitrani Rosenbaum S. GNE myopathy: two clusters with history and several founder mutations. J Neuromuscul Dis. 2015; 2:S73–S76. PMID: 27858758.
Article8. Malicdan MC, Noguchi S, Hayashi YK, Nonaka I, Nishino I. Prophylactic treatment with sialic acid metabolites precludes the development of the myopathic phenotype in the DMRV-hIBM mouse model. Nat Med. 2009; 15:690–695. PMID: 19448634.
Article9. Xu X, Wang AQ, Latham LL, Celeste F, Ciccone C, Malicdan MC, et al. Safety, pharmacokinetics and sialic acid production after oral administration of N-acetylmannosamine (ManNAc) to subjects with GNE myopathy. Mol Genet Metab. 2017; 122:126–134. PMID: 28641925.10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. PMID: 25741868.
Article11. Lu X, Pu C, Huang X, Liu J, Mao Y. Distal myopathy with rimmed vacuoles: clinical and muscle morphological characteristics and spectrum of GNE gene mutations in 53 Chinese patients. Neurol Res. 2011; 33:1025–1031. PMID: 22196754.
Article12. Kim BJ, Ki CS, Kim JW, Sung DH, Choi YC, Kim SH. Mutation analysis of the GNE gene in Korean patients with distal myopathy with rimmed vacuoles. J Hum Genet. 2006; 51:137–140. PMID: 16372135.
Article13. Sim JE, Park HJ, Shin HY, Nam TS, Kim SM, Choi YC. Clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy. Yonsei Med J. 2013; 54:578–582. PMID: 23549799.
Article14. Cho A, Hayashi YK, Monma K, Oya Y, Noguchi S, Nonaka I, et al. Mutation profile of the GNE gene in Japanese patients with distal myopathy with rimmed vacuoles (GNE myopathy). J Neurol Neurosurg Psychiatry. 2014; 85:914–917. PMID: 24027297.
Article15. Zhao J, Wang Z, Hong D, Lv H, Zhang W, Chen J, et al. Mutational spectrum and clinical features in 35 unrelated mainland Chinese patients with GNE myopathy. J Neurol Sci. 2015; 354:21–26. PMID: 25986339.
Article16. Mori-Yoshimura M, Hayashi YK, Yonemoto N, Nakamura H, Murata M, Takeda S, et al. Nationwide patient registry for GNE myopathy in Japan. Orphanet J Rare Dis. 2014; 9:150. PMID: 25303967.
Article17. Pogoryelova O, Cammish P, Mansbach H, Argov Z, Nishino I, Skrinar A, et al. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy disease monitoring program, registry portion. Neuromuscul Disord. 2018; 28:158–168. PMID: 29305133.
Article18. Mori-Yoshimura M, Okuma A, Oya Y, Fujimura-Kiyono C, Nakajima H, Matsuura K, et al. Clinicopathological features of centronuclear myopathy in Japanese populations harboring mutations in dynamin 2. Clin Neurol Neurosurg. 2012; 114:678–683. PMID: 22613877.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Progression of GNE Myopathy Based on the Patient-Reported Outcome
- Novel Mutation of the GNE Gene Presenting Atypical Mild Clinical Feature: A Korean Case Report
- A Case of GNE Myopathy Presenting a Rapid Deterioration during Pregnancy
- A Case of Nonaka Myopathy Confirmed by GNE Mutation
- Clinical Characteristics and Molecular Genetic Analysis of Korean Patients with GNE Myopathy
