A novel CHD7 mutation in an adolescent presenting with growth and pubertal delay
- Antoniou M
- Bouthors T
- Xu C
- Phan-Hug F
- Elowe-Gruau E
- Stoppa-Vaucher S
- van der Sloot A
- Acierno J
- Cassatella D
- Richard C
- Dwyer A
- Pitteloud
- Hauschild

- Affiliations
-
- 1Department of Pediatric Endocrinology and Diabetology, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland. Michael.Hauschild@chuv.ch
- 2Service of Endocrinology, Diabetes and Metabolism, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland.
- 3Institute for Research in Immunology and Cancer (IRIC), University of Montreal, Montreal, Canada.
- 4Otorhinolaryngology Service, Centre Hospitalier Universitaire Vaudois (CHUV), Lausanne, Switzerland.
- KMID: 2447376
- DOI: http://doi.org/10.6065/apem.2019.24.1.49
Abstract
- Mutations in the CHD7 gene, encoding for the chromodomain helicase DNA-binding protein 7, are found in approximately 60% of individuals with CHARGE syndrome (coloboma, heart defects, choanal atresia, retarded growth and development, genital hypoplasia, ear abnormalities and/or hearing loss). Herein, we present a clinical case of a 14-year-old male presenting for evaluation of poor growth and pubertal delay highlighting the diagnostic challenges of CHARGE syndrome. The patient was born full term and underwent surgery at 5 days of life for bilateral choanal atresia. Developmental milestones were normally achieved. At age 14 his height and weight were -2.04 and -1.74 standard deviation score respectively. He had anosmia as well as prepubertal testes and micropenis (4 cm×1 cm). The biological profile showed low basal serum testosterone and gonadotropins (testosterone, 0.2 nmol/L; luteinizing hormone, 0.5 U/L; follicle-stimulating hormone, 1.3 U/L), and otherwise normal pituitary function and normal imaging of the hypothalamic-pituitary area. The constellation of choanal atresia, anosmia, mild dysmorphic features, micropenis and delayed puberty were suggestive of CHARGE syndrome. Targeted genetic testing of CHD7 was performed revealing a de novo heterozygous CHD7 mutation (c.4234T>G [p.Tyr1412Asp]). Further paraclinical investigations confirmed CHARGE syndrome. Despite the presence of suggestive features, CHARGE syndrome remained undiagnosed in this patient until adolescence. Genetic testing helps clarify the phenotypic and genotypic spectrum to facilitate diagnosis, thus promoting optimal follow-up, treatment, and appropriate genetic counselling.
Keyword
MeSH Terms
-
Adolescent*
CHARGE Syndrome
Choanal Atresia
Diagnosis
Ear
Follicle Stimulating Hormone
Follow-Up Studies
Genetic Testing
Gonadotropins
Growth and Development
Hearing
Heart
Humans
Luteinizing Hormone
Male
Olfaction Disorders
Puberty, Delayed
Testis
Testosterone
Follicle Stimulating Hormone
Gonadotropins
Luteinizing Hormone
Testosterone
Figure
-
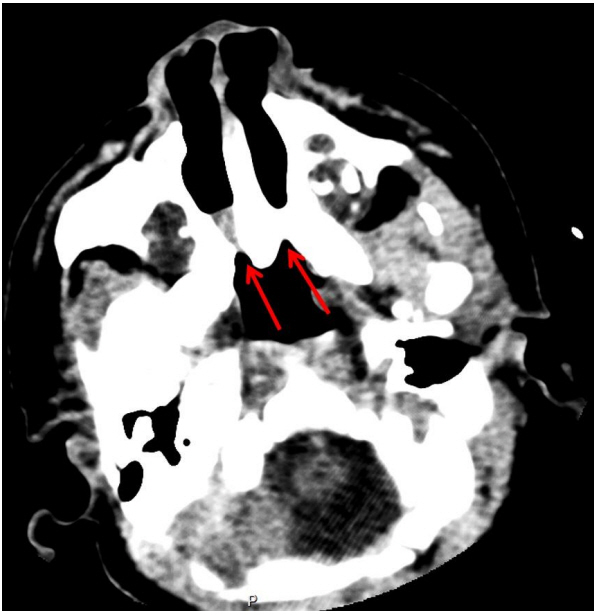
Fig. 1. Axial computed tomography scan at day 3 of life at the reference level of the pterygoid plates, showing bilateral choanal atresia (arrows).
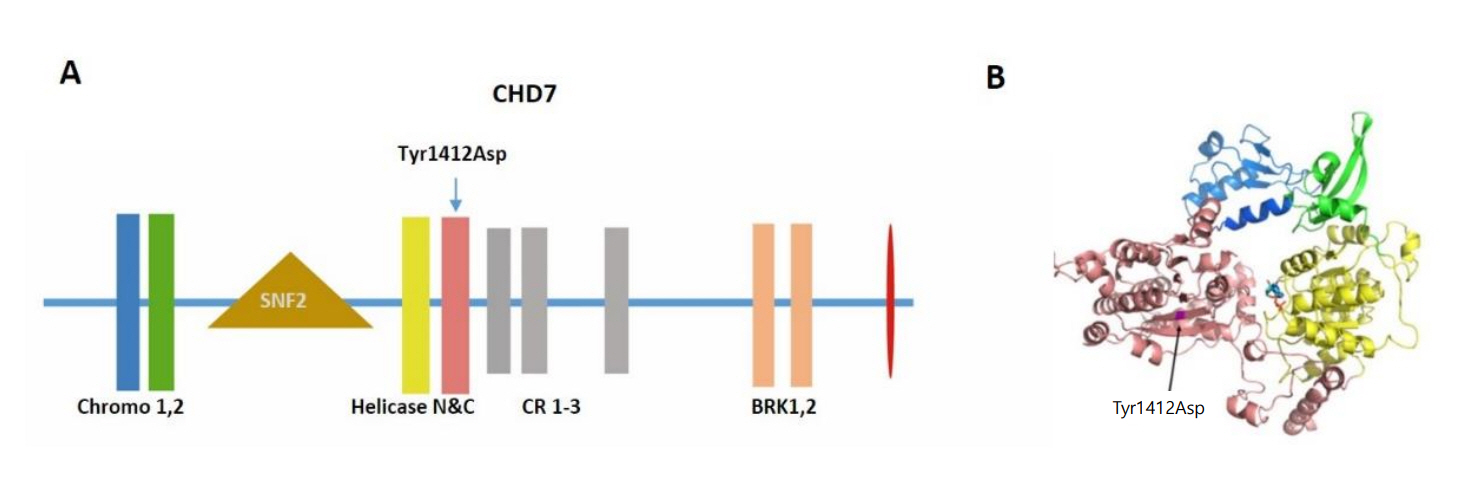
Fig. 2. (A) The schematic representation of CHD7 protein and the location of mutation Tyr1412Asp. The functional domains are indicated in the schema. (B) The structural modeling of chromodomains and helicase domains of CHD7 and the location of Try1412Asp. Tyr residue at position 1412 is located in the core of the helicase C domain. The change to an Asp is predicted to have a major detrimental effect on the structural stability of CHD7 as a large buried hydrophobic amino acid is replaced by a small charged hydrophilic one.

Fig. 3. (A-D) Imaging studies showing midline and intraauricular malformations. (A) Magnetic resonance imaging (MRI) (axial T1 with gadolinium) at 14 years of age, showing hypoplastic lateral and posterior semicircular canals (long arrows) and dysplastic vestibule (short arrow). (B) MRI at 14 years of age, showing Mondini dysplasia of the partition of the cochlea. (C) MRI at 14 years of age, showing decreased anterior pituitary volume (160 mm3). (D) MRI at 14 years of age, showing olfactory bulb aplasia (R) and hypoplasia (L). R, right; L, left.
Reference
-
References
1. Blake KD, Prasad C. CHARGE syndrome. Orphanet J Rare Dis. 2006; 1:34.
Article2. Bergman JE, de Ronde W, Jongmans MC, Wolffenbuttel BH, Drop SL, Hermus A, et al. The results of CHD7 analysis in clinically well-characterized patients with Kallmann syndrome. J Clin Endocrinol Metab. 2012; 97:E858–62.3. Verloes A. Updated diagnostic criteria for CHARGE syndrome: a proposal. Am J Med Genet A. 2005; 133A:306–8.
Article4. Jongmans MC, Admiraal RJ, van der Donk KP, Vissers LE, Baas AF, Kapusta L, et al. CHARGE syndrome: the phenotypic spectrum of mutations in the CHD7 gene. J Med Genet. 2006; 43:306–14.
Article5. van Ravenswaaij-Arts CM, Blake K, Hoefsloot L, Verloes A. Clinical utility gene card for: CHARGE syndrome - update 2015. Eur J Hum Genet. 2015; 23(11):https://doi.org/10.1038/ejhg.2015.15.
Article6. Basson MA, van Ravenswaaij-Arts C. Functional insights into chromatin remodelling from studies on CHARGE syndrome. Trends Genet. 2015; 31:600–11.
Article7. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J Med Genet. 2011; 48:334–42.
Article8. Hall JG, Froster-Iskenius UG, Allanson JE. Handbook of normal physical measurements. Oxford: Oxford Medical Publications;1989.9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–24.
Article10. Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017; 9:13.
Article11. Sinha V, Gurnani D, Modi NR, Barot DA, Maniyar HR, Pandey A. Choanal atresia: surgical management by Hegar's dilators. Indian J Otolaryngol Head Neck Surg. 2014; 66:272–5.
Article12. Burrow TA, Saal HM, de Alarcon A, Martin LJ, Cotton RT, Hopkin RJ. Characterization of congenital anomalies in individuals with choanal atresia. Arch Otolaryngol Head Neck Surg. 2009; 135:543–7.
Article13. Kim H, Park JH, Chung H, Han DH, Kim DY, Lee CH, et al. Clinical features and surgical outcomes of congenital choanal atresia: factors influencing success from 20-year review in an institute. Am J Otolaryngol. 2012; 33:308–12.
Article14. Hale CL, Niederriter AN, Green GE, Martin DM. Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A. 2016; 170A:344–54.15. Dwyer AA, Raivio T, Pitteloud N. Management of endocrine disease: reversible hypogonadotropic hypogonadism. Eur J Endocrinol. 2016; 174:R267–74.
Article16. Sanlaville D, Verloes A. CHARGE syndrome: an update. Eur J Hum Genet. 2007; 15:389–99.
Article17. Jongmans MC, van Ravenswaaij-Arts CM, Pitteloud N, Ogata T, Sato N, Claahsen-van der Grinten HL, et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome--the clinical overlap with CHARGE syndrome. Clin Genet. 2009; 75:65–71.
Article18. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism-- pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015; 11:547–64.
Article19. Laitinen EM, Tommiska J, Sane T, Vaaralahti K, Toppari J, Raivio T. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS One. 2012; 7:e39450.20. Dwyer AA, Phan-Hug F, Hauschild M, Elowe-Gruau E, Pitteloud N. Transition in endocrinology: hypogonadism in adolescence. Eur J Endocrinol. 2015; 173:R15–24.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Pubertal growth and epiphyseal fusion
- Identification of a novel mutation in the CHD7 gene in a patient with CHARGE syndrome
- Hypogonadotropic Hypogonadism and Abnormal Olfactory Bulb Development in CHARGE Syndrome with CHD7 Mutation
- A longitudinal study on the pubertal growth peak and maturity stages of the hand-wrist in malocclusion
- A case of CHARGE syndrome featuring immunodeficiency and hypocalcemia
