Functional Identification of Compound Heterozygous Mutations in the CYP17A1 Gene Resulting in Combined 17α-Hydroxylase/17,20-Lyase Deficiency
- Affiliations
-
- 1Department of Internal Medicine, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, Incheon, Korea. sungdaem@gmail.com
- 2Department of Laboratory Medicine, Incheon St. Mary's Hospital, College of Medicine, The Catholic University of Korea, Incheon, Korea.
- KMID: 2447032
- DOI: http://doi.org/10.3803/EnM.2018.33.3.413
Abstract
- BACKGROUND
We previously reported a patient with congenital adrenal hyperplasia (CAH) with compound heterozygous mutations in the cytochrome P450 17A1 (CYP17A1) gene. One allele had a p.His373Leu and the other a new p.Glu383fsX36 mutation. The aim of this study was to investigate the functional properties of a new allele present in a compound heterozygote of CYP17A1.
METHODS
To understand how p.His373Leu and p.Glu383fsX36 affect P450c17 enzymatic activity, wild type and mutant CYP17A1 cDNAs were cloned into flag-tagged pcDNA3 vector and introduced into human embryonic kidney cells 293T (HEK293T) cells. Protein expression levels of CYP17A1 were then analyzed. And the activities of 17α-hydroxylase and 17,20-lyase of CYP17A1 were evaluated by measuring the conversion of progesterone to 17α-hydroxyprogesterone and of 17α-hydroxypregnenolone to dehydroepiandrosterone, respectively. In addition a computer model was used to create the three-dimensional structure of the mutant CYP17A1 enzymes.
RESULTS
Production of the p.His373Leu mutant protein was significantly lower than that of the wild type protein, and the p.Glu383fsX36 protein was hardly produced. Similarly the enzymatic activity derived from the p.His373Leu mutant vector was significantly lower than that obtained from the wild type vector, and little activity was obtained from the p.Glu383fsX36 vector. Three-dimensional modeling of the enzyme showed that p.His373 was located in region important for heme-binding and proper folding. Neither the p.His373Leu nor the p.Glu383fsX36 mutant protein formed a heme-binding structure.
CONCLUSION
Enzyme activity measured in both mutants disappeared completely in both 17α-hydroxylase and 17,20-lyase. This result accounts for the clinical manifestations of the patient with the compound heterozygous CYP17A1 mutations.
Keyword
MeSH Terms
-
Adrenal Hyperplasia, Congenital
Alleles
Clone Cells
Computer Simulation
Cytochrome P-450 Enzyme System
Dehydroepiandrosterone
DNA, Complementary
Heterozygote
Humans
Kidney
Mutant Proteins
Progesterone
Steroid 17-alpha-Hydroxylase
Cytochrome P-450 Enzyme System
DNA, Complementary
Dehydroepiandrosterone
Mutant Proteins
Progesterone
Steroid 17-alpha-Hydroxylase
Figure
-
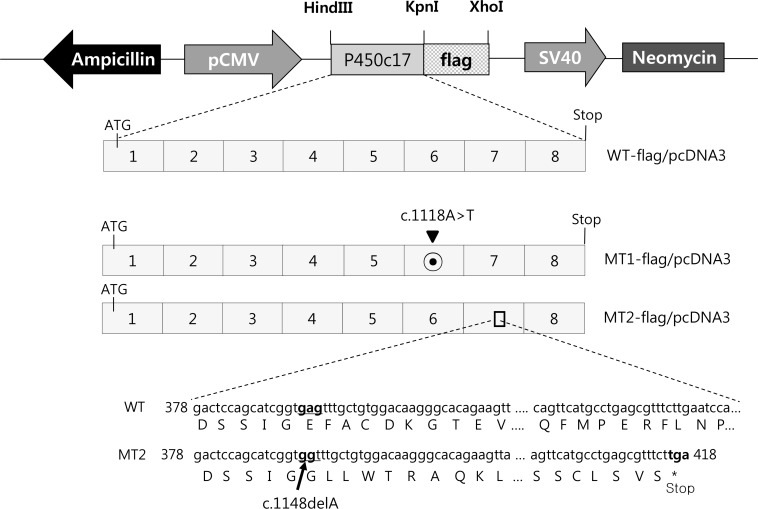
Fig. 1 Schematic representation of the constructs used to measure P450c17 enzyme activity produced in human embryonic kidney cells 293T (HEK293T) cells. The entire coding regions of c.1118A>T, c.1148delA, and wild type (WT) cytochrome P450 17A1 (CYP17A1) complementary DNAs (cDNAs) were subcloned into the HindIII and KpnI site of pcDNA3 vector with a C-terminal flag tag. The positions of restriction enzymes, start (ATG) and stop (tga) codons, and HindIII, KpnI, and XhoI sites, are shown. Exons are indicated with arabic numerals and the mutation sites (c.1118A>T and c.1148delA) are indicated. The 1 bp deletion (bp position 1148 in c.1148delA, indicated by the arrow) results in a shift of the reading frame and a termination codon after 36 amino acids. pCMV, promoter cytomegalovirus; SV40, simian virus 40; MT1, mutant type 1; MT2, mutant type 2.
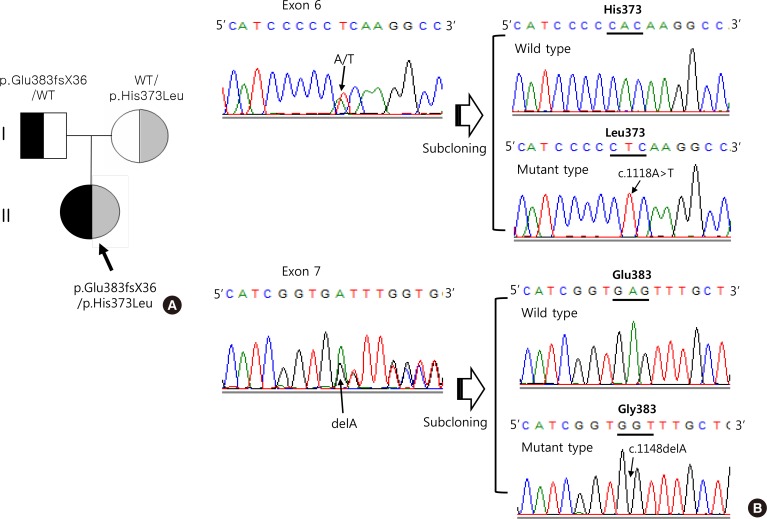
Fig. 2 The compound cytochrome P450 17A1 (CYP17A1) heterozygote. (A) Pedigree of 17α-hydroxylase/17,20-lyase deficient family. Individuals are represented as follows: male (square); females (circles). The black symbol represents the c.1148delA allele and the gray symbol represents the c.1118A>T allele. The arrow indicates the compound heterozygous proband. The proband inherited mutations c.1118A>T and c.1148delT from her mother and father, respectively. (B) Subcloning of the compound heterozygous CYP17A1 gene. Polymerase chain reaction products of RNA from the proband's leucocytes were subcloned and sequenced to confirm heterozygous mutations. The c.1148delA mutation was found to be in exon 7 and the c.1118A>T mutation in exon 6. WT, wild type; A/T, A and T.
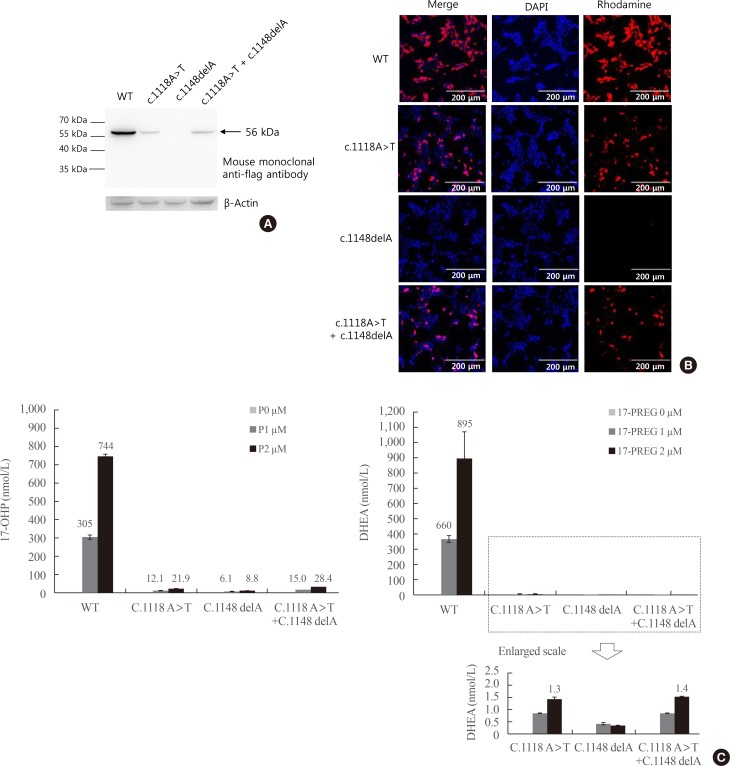
Fig. 3 Expression and enzyme activities of P450c17 protein produced by cytochrome P450 17A1 (CYP17A1) complementary DNA (cDNA) overexpressed in human embryonic kidney cells 293T (HEK293T) cells. (A, B) Analysis of P450c17 proteins by Western blotting (WB) and immunofluorescence, respectively. Expression of c.1118A>T and the doubly transfected clone (p.H373L and p.E383fsX36) in HEK293T cells is significantly lower than that of wild type (WT) and no expression of c.1148delA mutant is detected. (C) Enzyme activity. Production of 17α-hydroxyprogesterone (17-OHP) in the presence of various concentrations of progesterone (P) (0, 1.0, and 2.0 µM) and production of dehydroepiandrosterone (DHEA) in the presence of various concentration of 17α-hydroxypregnenolone (17-PREG) (0, 1.0, and 2.0 µM). HEK293T cells were transfected with WT, c.1118A>T, c.1148delA, and co-transfection. Compared to the enzymatic activity against WT (744 nmol/L), the 17α-hydroxylase activity for the p.His373Leu mutant was reduced to 21.9 nmol/L and the 17,20-lyase activity for the p.Glu383fsX36 mutant was almost zero (1.3 nmol/L). Data are mean values of P450c17 activity assayed in triplicate. Data are expressed as mean±SD (n=3). DAPI, 4',6-diamidino-2-phenylindole.
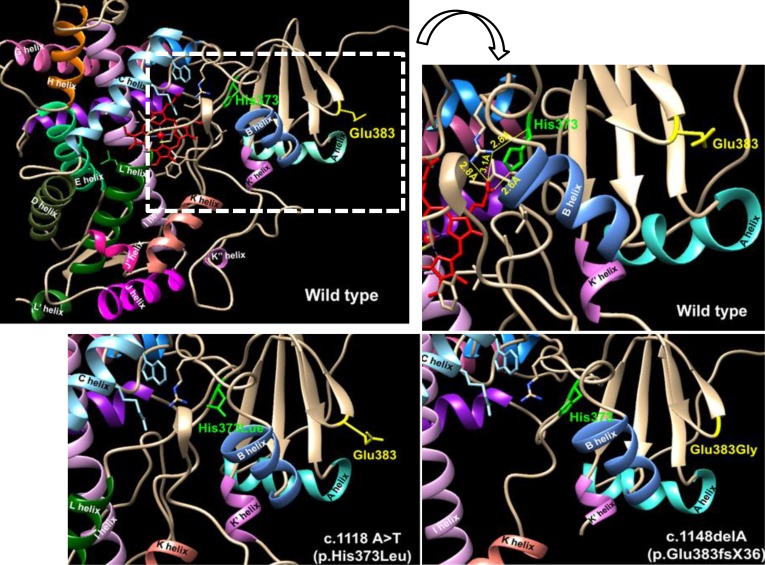
Fig. 4 Computer model of cytochrome P450 17A1 (CYP17A1) (PDB ID; 3SWZ). A, a ribbon diagram of the protein backbone, with heme (red) and highlighting p.His373, p.Glu383, p.His373Leu, p.Glu383Gly. Also shown are residues p.His373 and p.His373Leu in green and p.Glu383 and p.Glu383Gly in yellow. Substitution of His by Leu at amino acid position 373 results in loss of catalytic activity. Mutation of the His-deficient mutant at position 373 or substitution of Glu with Gly at position 383 prevents the incorporation of a heme group, rendering the mutant enzyme inactive. The imidazole side chain of His is a common ligand for the metal protein and is part of the catalytic site of the enzyme. His can participate in acidic catalysis by proton and non-proton states, but Leu does not bind to heme because there is no imidazole branch. Images were created in Swiss PDB viewer (Swiss Institute of Bioinformatics) and UCSF Chimera version 1.11.2 (University of California).
Reference
-
1. Zhang M, Sun S, Liu Y, Zhang H, Jiao Y, Wang W, et al. New, recurrent, and prevalent mutations: clinical and molecular characterization of 26 Chinese patients with 17alpha-hydroxylase/17,20-lyase deficiency. J Steroid Biochem Mol Biol. 2015; 150:11–16. PMID: 25697092.
Article2. Yamaguchi H, Nakazato M, Miyazato M, Toshimori H, Oki S, Shimizu K, et al. Identification of a novel splicing mutation and 1-bp deletion in the 17alpha-hydroxylase gene of Japanese patients with 17alpha-hydroxylase deficiency. Hum Genet. 1998; 102:635–639. PMID: 9703423.3. Yanase T, Simpson ER, Waterman MR. 17 Alpha-hydroxy-lase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991; 12:91–108. PMID: 2026124.4. Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017; 165(Pt A):71–78. PMID: 26862015.
Article5. Jameson JL, De Groot LJ. Endocrinology: adult and pediatric. 7th ed. Philadelphia: Elsevier Saunders;2016. p. 1810–1832.6. Biglieri EG, Herron MA, Brust N. 17-Hydroxylation deficiency in man. J Clin Invest. 1966; 45:1946–1954. PMID: 4288776.
Article7. Han B, Liu W, Zuo CL, Zhu H, Li L, Xu C, et al. Identifying a novel mutation of CYP17A1 gene from five Chinese 17α-hydroxylase/17, 20-lyase deficiency patients. Gene. 2013; 516:345–350. PMID: 23291414.
Article8. Lee ES, Kim M, Moon S, Jekarl DW, Lee S, Kim Y, et al. A new compound heterozygous mutation in the CYP17A1 gene in a female with 17α-hydroxylase/17,20-lyase deficiency. Gynecol Endocrinol. 2013; 29:720–723. PMID: 23772786.9. Moreira AC, Leal AM, Castro M. Characterization of adrenocorticotropin secretion in a patient with 17 alpha-hydroxylase deficiency. J Clin Endocrinol Metab. 1990; 71:86–91. PMID: 2164530.10. Heremans GF, Moolenaar AJ, van Gelderen HH. Female phenotype in a male child due to 17-alpha-hydroxylase deficiency. Arch Dis Child. 1976; 51:721–723. PMID: 999330.
Article11. Gupta MK, Geller DH, Auchus RJ. Pitfalls in characterizing P450c17 mutations associated with isolated 17,20-lyase deficiency. J Clin Endocrinol Metab. 2001; 86:4416–4423. PMID: 11549685.
Article12. Patocs A, Liko I, Varga I, Gergics P, Boros A, Futo L, et al. Novel mutation of the CYP17 gene in two unrelated patients with combined 17alpha-hydroxylase/17,20-lyase deficiency: demonstration of absent enzyme activity by expressing the mutant CYP17 gene and by three-dimensional modeling. J Steroid Biochem Mol Biol. 2005; 97:257–265. PMID: 16176874.13. Bao X, Ding H, Xu Y, Cui G, He Y, Yu X, et al. Prevalence of common mutations in the CYP17A1 gene in Chinese Han population. Clin Chim Acta. 2011; 412:1240–1243. PMID: 21420394.
Article14. Kim YM, Kang M, Choi JH, Lee BH, Kim GH, Ohn JH, et al. A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase deficiency, a case series of such mutations among Koreans and functional characteristics of a novel mutation. Metabolism. 2014; 63:42–49. PMID: 24140098.
Article15. Tian Q, Yao F, Sha G, Huang S, Tseng H, Schindler AE. Genotyping of a Chinese family with 46,XX and 46,XY 17-hydroxylase deficiency. Gynecol Endocrinol. 2009; 25:485–490. PMID: 19499410.
Article16. Melmed S, Polonsky KS, Larsen PR, Kronnenberg HM. Williams textbook of endocrinology. 13th ed. Philadelphia: Elsevier Saunders;2016. p. 540–541.17. Costa-Santos M, Kater CE, Auchus RJ;. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004; 89:49–60. PMID: 14715827.18. Geller DH, Auchus RJ, Miller WL. P450c17 mutations R347H and R358Q selectively disrupt 17,20-lyase activity by disrupting interactions with P450 oxidoreductase and cytochrome b5. Mol Endocrinol. 1999; 13:167–175. PMID: 9892022.
Article19. Okabayashi T, Shima Y, Sumiyoshi T, Kozuki A, Ito S, Ogawa Y, et al. Diagnosis and management of insulinoma. World J Gastroenterol. 2013; 19:829–837. PMID: 23430217.
Article20. Kater CE, Biglieri EG. Disorders of steroid 17 alpha-hydroxylase deficiency. Endocrinol Metab Clin North Am. 1994; 23:341–357. PMID: 8070426.21. Geller DH, Auchus RJ, Mendonca BB, Miller WL. The genetic and functional basis of isolated 17,20-lyase deficiency. Nat Genet. 1997; 17:201–205. PMID: 9326943.
Article22. Sherbet DP, Tiosano D, Kwist KM, Hochberg Z, Auchus RJ. CYP17 mutation E305G causes isolated 17,20-lyase deficiency by selectively altering substrate binding. J Biol Chem. 2003; 278:48563–48569. PMID: 14504283.
Article23. Tiosano D, Knopf C, Koren I, Levanon N, Hartmann MF, Hochberg Z, et al. Metabolic evidence for impaired 17alpha-hydroxylase activity in a kindred bearing the E305G mutation for isolate 17,20-lyase activity. Eur J Endocrinol. 2008; 158:385–392. PMID: 18299473.24. Biglieri EG. Mechanisms establishing the mineralocorticoid hormone patterns in the 17 alpha-hydroxylase deficiency syndrome. J Steroid Biochem. 1979; 11(1B):653–657. PMID: 226795.25. Dean HJ, Shackleton CH, Winter JS. Diagnosis and natural history of 17-hydroxylase deficiency in a newborn male. J Clin Endocrinol Metab. 1984; 59:513–520. PMID: 6086702.
Article26. Morimoto I, Maeda R, Izumi M, Ishimaru T, Nishimori I, Nagataki S. An autopsy case of 17 alpha-hydroxylase deficiency with malignant hypertension. J Clin Endocrinol Metab. 1983; 56:915–919. PMID: 6300176.27. Lee MH, Park SW, Yoon TK, Shim SH. Homozygous CYP17A1 mutation (H373L) identified in a 46,XX female with combined 17α-hydroxylase/17,20-lyase deficiency. Gynecol Endocrinol. 2012; 28:573–576. PMID: 22452398.28. Katsumata N, Ogawa E, Fujiwara I, Fujikura K. Novel CYP17A1 mutation in a Japanese patient with combined 17alpha-hydroxylase/17,20-lyase deficiency. Metabolism. 2010; 59:275–278. PMID: 19793597.29. Sahakitrungruang T, Tee MK, Speiser PW, Miller WL. Novel P450c17 mutation H373D causing combined 17alpha-hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab. 2009; 94:3089–3092. PMID: 19470621.30. Qiao J, Hu RM, Peng YD, Song HD, Peng YW, Gao GF, et al. A complex heterozygous mutation of His373Leu and Asp487-Ser488-Phe489 deletion in human cytochrome P450c17 causes 17alpha-hydroxylase/17,20-lyase deficiency in three Chinese sisters. Mol Cell Endocrinol. 2003; 201:189–195. PMID: 12706306.31. Katsumata N, Satoh M, Mikami A, Mikami S, Nagashima-Miyokawa A, Sato N, et al. New compound heterozygous mutation in the CYP17 gene in a 46,XY girl with 17 alpha-hydroxylase/17,20-lyase deficiency. Horm Res. 2001; 55:141–146. PMID: 11549876.32. Shin DH, Yu SH, Choi YM, Kim JG, Kim SW, Shin CS, et al. Six cases of congenital adrenal hyperplasia that were due to 17alpha-hydroxylase/17,20-lyase deficiency. J Korean Endocr Soc. 2009; 24:109–115.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Two cases of 17α-hydroxylase/17,20-lyase deficiency caused by the CYP17A1 mutation
- Prevalence of CYP17A1 gene mutations in 17α-hydroxylase deficiency in the Chinese Han population
- Two Cases of Simple Virilizing Congenital Adrenal Hyperplasia with Compound Heterozygous Mutations of CYP21 Gene
- Limited Expression of Cytochrome P450 17alpha-Hydroxylase/17,20-Lyase in Prostate Cancer Cell Lines
- A case of 17 alpha-hydroxylase deficiency
