Recent Topics in Fibrodysplasia Ossificans Progressiva
- Affiliations
-
- 1Division of Pathophysiology, Research Center for Genomic Medicine, Saitama Medical University, Saitama, Japan. katagiri@saitama-med.ac.jp
- 2Project of Clinical and Basic Research for FOP, Saitama Medical University, Saitama, Japan.
- KMID: 2447022
- DOI: http://doi.org/10.3803/EnM.2018.33.3.331
Abstract
- Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disease that is characterized by the formation of heterotopic bone tissues in soft tissues, such as skeletal muscle, ligament, and tendon. It is difficult to remove such heterotopic bones via internal medicine or invasive procedures. The identification of activin A receptor, type I (ACVR1)/ALK2 gene mutations associated with FOP has allowed the genetic diagnosis of FOP. The ACVR1/ALK2 gene encodes the ALK2 protein, which is a transmembrane kinase receptor in the transforming growth factor-β family. The relevant mutations activate intracellular signaling in vitro and induce heterotopic bone formation in vivo. Activin A is a potential ligand that activates mutant ALK2 but not wild-type ALK2. Various types of small chemical and biological inhibitors of ALK2 signaling have been developed to establish treatments for FOP. Some of these are in clinical trials in patients with FOP.
MeSH Terms
Figure
-

Fig. 1 Schematic representation of the relationship between the activin A receptor, type I (ACVR1)/ALK2 gene, complementary DNA (cDNA) and protein. The ACVR1/ALK2 gene consist of 9 coding exons (Ex.) (black boxes). The ACVR1/ALK2 cDNA (1,530 bp) encodes a protein with 509 amino acids (a. a.). Mutations associated with fibrodysplasia ossificans progressiva are shown in the figure. The positions of the mutations in the cDNA and protein are indicated by numbers that begin from the adenine of the first ATG codon and Met residue, respectively. TGA, stop codon; SP, signal peptide; TM, transmembrane domain; GS, glycine/serine-rich domain; Ser/Thr kinase, serine/threonine kinase domain.
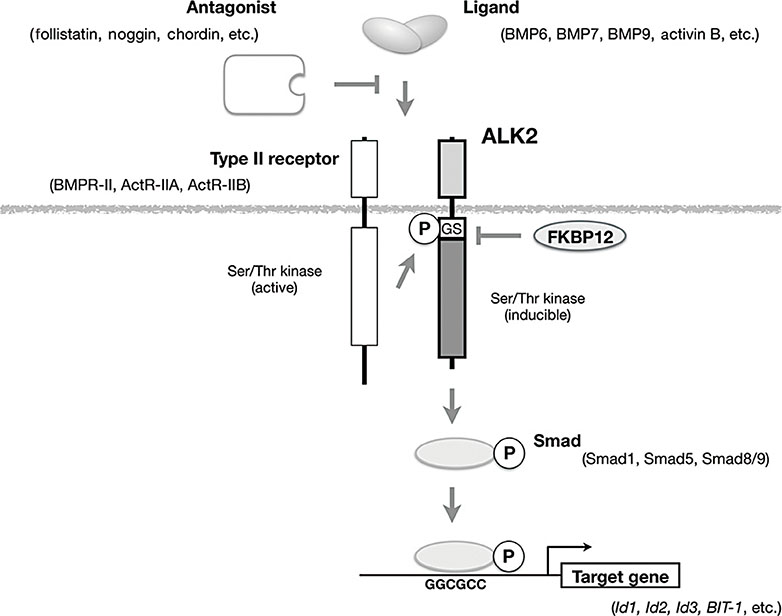
Fig. 2 Schematic representation of signal transduction by ALK2 in response to ligand binding. ALK2 binds to a transforming growth factor-β family ligand, such as bone morphogenetic protein 6 (BMP6), BMP7, and BMP9, and acts as a type I receptor in co-operation with one of the type II receptors (BMP receptor type II [BMPR-II], activin receptor type IIA [ActR-IIA], and activin receptor type IIB [ActR-IIB]). Antagonists, such as follistatin, noggin, and chordin, directly bind to the ligand and prevent it from binding to receptors. Type II receptors are constitutively active kinases that phosphorylate the glycine/serine-rich domain (GS) domain of ALK2 to activate kinase activity. Activated ALK2 phosphorylates downstream substrates, such as Smad1, Smad5, and Smad8/9, and then binds to specific DNA sequences to regulate the transcription of its target genes. Ser/Thr, serine/threonine; P, phosphorylation; FKBP12, 12 kDa FK506-binding protein; Id1, inhibitor of DNA binding 1; BIT-1, BMP-inducible transcript-1.
Reference
-
1. Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech. 2012; 5:756–762.
Article2. Katagiri T. Heterotopic bone formation induced by bone morphogenetic protein signaling: fibrodysplasia ossificans progressiva. J Oral Biosci. 2010; 52:33–41.
Article3. Katagiri T. A door opens for fibrodysplasia ossificans progressiva. Trends Biochem Sci. 2016; 41:119–121.
Article4. Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med. 1996; 335:555–561.
Article5. Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005; 116:e654–e661.
Article6. Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006; 38:525–527.
Article7. Urist MR. Bone: formation by autoinduction. Science. 1965; 150:893–899.
Article8. Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, et al. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am. 1993; 75:215–219.
Article9. Zaghloul KA, Heuer GG, Guttenberg MD, Shore EM, Kaplan FS, Storm PB. Lumbar puncture and surgical intervention in a child with undiagnosed fibrodysplasia ossificans progressiva. J Neurosurg Pediatr. 2008; 1:91–94.
Article10. Nakashima Y, Haga N, Kitoh H, Kamizono J, Tozawa K, Katagiri T, et al. Deformity of the great toe in fibrodysplasia ossificans progressiva. J Orthop Sci. 2010; 15:804–809.
Article11. Kaplan FS, Xu M, Glaser DL, Collins F, Connor M, Kitterman J, et al. Early diagnosis of fibrodysplasia ossificans progressiva. Pediatrics. 2008; 121:e1295–e1300.
Article12. Pignolo RJ, Bedford-Gay C, Liljesthrom M, Durbin-Johnson BP, Shore EM, Rocke DM, et al. The natural history of flare-ups in fibrodysplasia ossificans progressiva (FOP): a comprehensive global assessment. J Bone Miner Res. 2016; 31:650–656.
Article13. Feldman G, Li M, Martin S, Urbanek M, Urtizberea JA, Fardeau M, et al. Fibrodysplasia ossificans progressiva, a heritable disorder of severe heterotopic ossification, maps to human chromosome 4q27-31. Am J Hum Genet. 2000; 66:128–135.
Article14. Lucotte G, Bathelier C, Mercier G, Gerard N, Lenoir G, Semonin O, et al. Fibrodysplasia Ossificans Progressiva Consortium. Localization of the gene for fibrodysplasia ossificans progressiva (FOP) to chromosome 17q21-22. Genet Couns. 2000; 11:329–334.15. Xu MQ, Feldman G, Le Merrer M, Shugart YY, Glaser DL, Urtizberea JA, et al. Linkage exclusion and mutational analysis of the noggin gene in patients with fibrodysplasia ossificans progressiva (FOP). Clin Genet. 2000; 58:291–298.16. Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994; 370:341–347.17. Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002; 296:1646–1647.
Article18. Nojima J, Kanomata K, Takada Y, Fukuda T, Kokabu S, Ohte S, et al. Dual roles of Smad proteins in the conversion from myoblasts to osteoblastic cells by bone morphogenetic proteins. J Biol Chem. 2010; 285:15577–15586.
Article19. Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep. 2014; 4:7596.
Article20. Katagiri T, Imada M, Yanai T, Suda T, Takahashi N, Kamijo R. Identification of a BMP-responsive element in Id1, the gene for inhibition of myogenesis. Genes Cells. 2002; 7:949–960.21. Shin M, Ohte S, Fukuda T, Sasanuma H, Yoneyama K, Kokabu S, et al. Identification of a novel bone morphogenetic protein (BMP)-inducible transcript, BMP-inducible transcript-1, by utilizing the conserved BMP-responsive elements in the Id genes. J Bone Miner Metab. 2013; 31:34–43.
Article22. Fukuda T, Kanomata K, Nojima J, Kokabu S, Akita M, Ikebuchi K, et al. A unique mutation of ALK2, G356D, found in a patient with fibrodysplasia ossificans progressiva is a moderately activated BMP type I receptor. Biochem Biophys Res Commun. 2008; 377:905–909.
Article23. Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem. 2009; 284:7149–7156.
Article24. Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009; 119:3462–3472.
Article25. Fujimoto M, Ohte S, Osawa K, Miyamoto A, Tsukamoto S, Mizuta T, et al. Mutant activin-like kinase 2 in fibrodysplasia ossificans progressiva are activated via T203 by BMP type II receptors. Mol Endocrinol. 2015; 29:140–152.
Article26. Wang T, Li BY, Danielson PD, Shah PC, Rockwell S, Lechleider RJ, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996; 86:435–444.27. Machiya A, Tsukamoto S, Ohte S, Kuratani M, Fujimoto M, Kumagai K, et al. Effects of FKBP12 and type II BMP receptors on signal transduction by ALK2 activating mutations associated with genetic disorders. Bone. 2018; 111:101–108.
Article28. Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009; 30:379–390.
Article29. Gregson CL, Hollingworth P, Williams M, Petrie KA, Bullock AN, Brown MA, et al. A novel ACVR1 mutation in the glycine/serine-rich domain found in the most benign case of a fibrodysplasia ossificans progressiva variant reported to date. Bone. 2011; 48:654–658.
Article30. Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. J Bone Miner Res. 2012; 27:1746–1756.31. Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Sci Transl Med. 2015; 7:303ra137.32. Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proc Natl Acad Sci U S A. 2015; 112:15438–15443.
Article33. Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, et al. Identification of progenitor cells that contribute to heterotopic skeletogenesis. J Bone Joint Surg Am. 2009; 91:652–663.
Article34. Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nat Med. 2010; 16:1400–1406.
Article35. Wosczyna MN, Biswas AA, Cogswell CA, Goldhamer DJ. Multipotent progenitors resident in the skeletal muscle interstitium exhibit robust BMP-dependent osteogenic activity and mediate heterotopic ossification. J Bone Miner Res. 2012; 27:1004–1017.
Article36. Dey D, Bagarova J, Hatsell SJ, Armstrong KA, Huang L, Ermann J, et al. Two tissue-resident progenitor lineages drive distinct phenotypes of heterotopic ossification. Sci Transl Med. 2016; 8:366ra163.
Article37. Lees-Shepard JB, Yamamoto M, Biswas AA, Stoessel SJ, Nicholas SE, Cogswell CA, et al. Activin-dependent signaling in fibro/adipogenic progenitors causes fibrodysplasia ossificans progressiva. Nat Commun. 2018; 9:471.
Article38. Upadhyay J, Xie L, Huang L, Das N, Stewart RC, Lyon MC, et al. The expansion of heterotopic bone in fibrodysplasia ossificans progressiva is activin a-dependent. J Bone Miner Res. 2017; 32:2489–2499.
Article39. Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008; 4:33–41.
Article40. Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008; 14:1363–1369.41. Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, et al. In vivo structure-activity relationship study of dorsomorphin analogues identifies selective VEGF and BMP inhibitors. ACS Chem Biol. 2010; 5:245–253.
Article42. Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol. 2013; 8:1291–1302.
Article43. Sanvitale CE, Kerr G, Chaikuad A, Ramel MC, Mohedas AH, Reichert S, et al. A new class of small molecule inhibitor of BMP signaling. PLoS One. 2013; 8:e62721.
Article44. Engers DW, Frist AY, Lindsley CW, Hong CC, Hopkins CR. Synthesis and structure-activity relationships of a novel and selective bone morphogenetic protein receptor (BMP) inhibitor derived from the pyrazolo[1.5-a]pyrimidine scaffold of dorsomorphin: the discovery of ML347 as an ALK2 versus ALK3 selective MLPCN probe. Bioorg Med Chem Lett. 2013; 23:3248–3252.
Article45. Takahashi M, Katagiri T, Furuya H, Hohjoh H. Disease-causing allele-specific silencing against the ALK2 mutants, R206H and G356D, in fibrodysplasia ossificans progressiva. Gene Ther. 2012; 19:781–785.
Article46. Kaplan J, Kaplan FS, Shore EM. Restoration of normal BMP signaling levels and osteogenic differentiation in FOP mesenchymal progenitor cells by mutant allele-specific targeting. Gene Ther. 2012; 19:786–790.
Article47. Shi S, Cai J, de Gorter DJ, Sanchez-Duffhues G, Kemaladewi DU, Hoogaars WM, et al. Antisense-oligonucleotide mediated exon skipping in activin-receptor-like kinase 2: inhibiting the receptor that is overactive in fibrodysplasia ossificans progressiva. PLoS One. 2013; 8:e69096.
Article48. Shimono K, Morrison TN, Tung WE, Chandraratna RA, Williams JA, Iwamoto M, et al. Inhibition of ectopic bone formation by a selective retinoic acid receptor alpha-agonist: a new therapy for heterotopic ossification? J Orthop Res. 2010; 28:271–277.49. Shimono K, Tung WE, Macolino C, Chi AH, Didizian JH, Mundy C, et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat Med. 2011; 17:454–460.
Article50. Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al. Palovarotene inhibits heterotopic ossification and maintains limb mobility and growth in mice with the human ACVR1(R206H) fibrodysplasia ossificans progressiva (FOP) mutation. J Bone Miner Res. 2016; 31:1666–1675.51. Hino K, Horigome K, Nishio M, Komura S, Nagata S, Zhao C, et al. Activin-A enhances mTOR signaling to promote aberrant chondrogenesis in fibrodysplasia ossificans progressiva. J Clin Invest. 2017; 127:3339–3352.
Article52. Fukuda T, Uchida R, Inoue H, Ohte S, Yamazaki H, Matsuda D, et al. Fungal pyrrolidine-containing metabolites inhibit alkaline phosphatase activity in bone morphogenetic protein-stimulated myoblastoma cells. Acta Pharm Sin B. 2012; 2:23–27.
Article53. Fukuda T, Uchida R, Ohte S, Inoue H, Yamazaki H, Matsuda D, et al. Trichocyalides A and B, new inhibitors of alkaline phosphatase activity in bone morphogenetic protein-stimulated myoblasts, produced by Trichoderma sp. FKI-5513. J Antibiot (Tokyo). 2012; 65:565–569.
Article54. Yamamoto R, Matsushita M, Kitoh H, Masuda A, Ito M, Katagiri T, et al. Clinically applicable antianginal agents suppress osteoblastic transformation of myogenic cells and heterotopic ossifications in mice. J Bone Miner Metab. 2013; 31:26–33.
Article55. Uchida R, Nakai M, Ohte S, Onaka H, Katagiri T, Tomoda H. 5-Prenyltryptophol, a new inhibitor of bone morphogenetic protein-induced alkaline phosphatase expression in myoblasts, produced by Streptomyces colinus subsp. Albescens HEK608. J Antibiot (Tokyo). 2014; 67:589–591.
Article56. Uchida R, Lee D, Suwa I, Ohtawa M, Watanabe N, Demachi A, et al. Scopranones with two atypical scooplike moieties produced by streptomyces sp. BYK-11038. Org Lett. 2017; 19:5980–5983.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Head and neck manifestations of fibrodysplasia ossificans progressiva: Clinical and imaging findings in 2 cases
- Myositis Ossificans Progressiva: A Case Report
- Genetic Transmission of Fibrodysplasia Ossificans Progressiva: Report of Two Cases in a Family
- Myositis ossificans progressiva
- 3 Cases Report of Myositis Ossificans Progressiva
