Orthopedic Manifestations of Type I Camurati-Engelmann Disease
- Affiliations
-
- 1Division of Pediatric Orthopedics, Seoul National University Children's Hospital, Seoul, Korea. tjcho@snu.ac.kr
- 2Department of Traumatology and Orthopedics, Children Traumatology and Orthopedics, Neurosurgery and Children Neurosurgery, ashkent Pediatric Medical Institute, Tashkent, Uzbekistan.
- 3Department of Orthopedic Surgery, Korea University Anam Hospital, Seoul, Korea.
- 4Department of Orthopedic Surgery, Seoul National University Bundang Hospital, Seongnam, Korea.
- 5Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea.
- 6Department of Radiology, Woorisoa Children's Hospital, Seoul, Korea.
- KMID: 2412312
- DOI: http://doi.org/10.4055/cios.2017.9.1.109
Abstract
- BACKGROUND
Camurati-Engelmann disease (CED) is a rare genetic skeletal disorder characterized by limb pain, muscle emaciation and weakness, and cortical thickening of the diaphysis of long bones. It is caused by mutations in the transforming growth factor beta 1 (TGFB1) (type I) or other unknown gene(s) (type II). We present 8 consecutive patients with type I CED.
METHODS
We retrospectively reviewed medical records and radiographs of type I CED patients with special reference to the mode of presentation, process of diagnostic work-up, and disease course. They were 4 sporadic patients, and two pairs of mother and son.
RESULTS
We categorized the mode of presentation into three groups. Group I had 4 patients who mainly presented with motor disturbances in young age. They drew medical attention for waddling gait, awkward ambulation or running, difficulty in going upstairs, or a positive Gower's sign at age 4 to 6 years. Subsequent development of limb pain and radiographic abnormality led to the diagnosis of CED at age 6 to 29 years. Group II had 3 patients who mainly presented with limb pain at age 15, 20, and 54 years, respectively. Radiographic evaluation and molecular genetic test led to the diagnosis of CED. The remaining 1 patient (group III) was asymptomatic until age 9 years when bony lesions at the tibiae were found incidentally. For the last 10 years, he intermittently complained of leg pain in the morning or after sports activities, which did not interfere with daily life. All the patients in group I showed a body mass index in the underweight range (< 18.4 kg/m²). At the latest follow-up, 4 patients in groups I and II required medication for the limb pain.
CONCLUSIONS
CED presents with a wide range of severity. Awareness of this rare disease entity may be the key to timely correct diagnosis. This disease entity should be considered in the differential diagnosis of limb pain or motor disturbance in children to avoid unnecessary diagnostic work-up.
MeSH Terms
-
Adolescent
Adult
Analgesics/therapeutic use
Camurati-Engelmann Syndrome/*complications/*diagnostic imaging/genetics/physiopathology
Child
Child, Preschool
Female
*Gait
Genetic Testing
Humans
Male
Middle Aged
Mobility Limitation
Musculoskeletal Pain/drug therapy/*etiology
Pain Measurement
Retrospective Studies
Severity of Illness Index
Stair Climbing
Transforming Growth Factor beta1/genetics
Young Adult
Analgesics
Transforming Growth Factor beta1
Figure
-
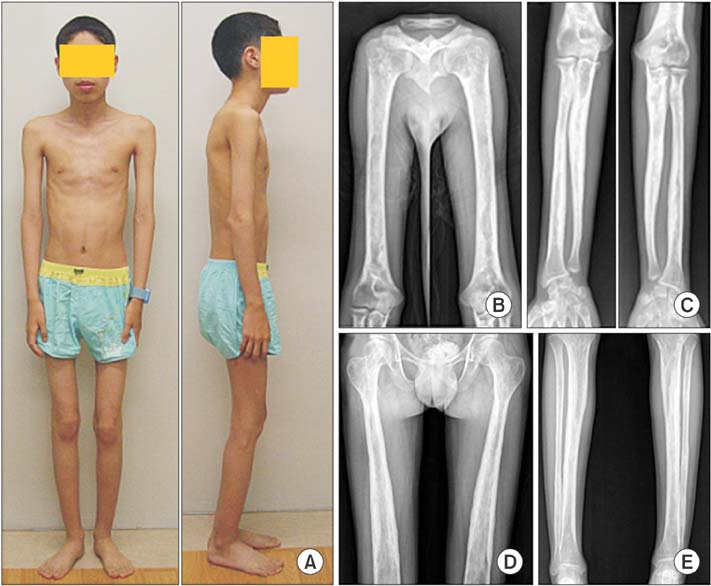
Fig. 1 Photographs and radiographs of a 19-year-old man (patient 2) show slim body habitus (A) and cortical hyperostosis of the long bones of the upper (B, C) and lower (D, E) extremities.
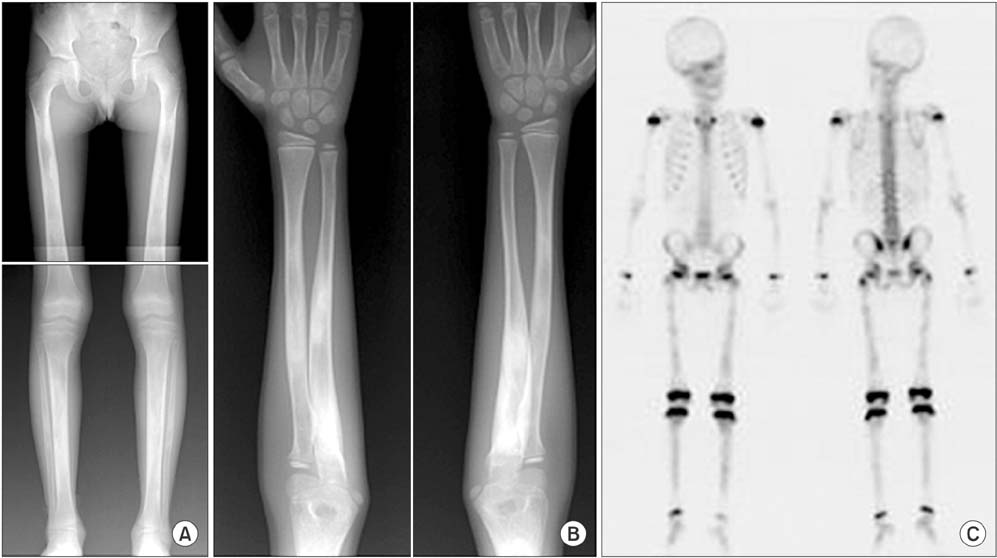
Fig. 2 Radiographs of a 10-year-old boy (patient 3) show symmetrical cortical hyperostosis of the long bones (A, B), and scintigraphy shows moderate hot uptake in the diaphyses of femora, tibiae and both forearm bones (C).

Fig. 3 Radiographs of a 57-year-old woman (patient 7) show marked deformities of the foot and ankle (A, B), hyperostosis of the skull (C), and anterior dislocation of the radial head (D).
Cited by 1 articles
-
Regarding Camurati-Engelmann Disease: To the Editor
Melissa Machado Viana, Sabrina Versuti Nunes, Davi Coutinho F. Fernandes Gomes, Marco Antônio Percope de Andrade, Marcos José Burle de Aguiar
Clin Orthop Surg. 2018;10(1):116-117. doi: 10.4055/cios.2018.10.1.116.
Reference
-
1. Grey AC, Wallace R, Crone M. Engelmann's disease: a 45-year follow-up. J Bone Joint Surg Br. 1996; 78(3):488–491.2. Brat HG, Hamoir X, Matthijs P, Lambin P, Van Campenhoudt M. Camurati-Engelmann disease: a late and sporadic case with metaphyseal involvement. Eur Radiol. 1999; 9(1):159–162.
Article3. Janssens K, Vanhoenacker F, Bonduelle M, et al. Camurati-Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006; 43(1):1–11.
Article4. Kinoshita A, Saito T, Tomita H, et al. Domain-specific mutations in TGFB1 result in Camurati-Engelmann disease. Nat Genet. 2000; 26(1):19–20.
Article5. Nishimura G, Nishimura H, Tanaka Y, et al. Camurati-Engelmann disease type II: progressive diaphyseal dysplasia with striations of the bones. Am J Med Genet. 2002; 107(1):5–11.
Article6. Lucke T, Illsinger S, Das AM, Schirg E, Hartmann H. Pitfalls in paediatric gait disturbances: painless bone diseases. Eur J Pediatr. 2006; 165(12):909–912.7. Ferraz-Amaro I, Delgado-Frias E, Hernandez-Hernandez V, Diaz-Gonzalez F. A woman with joint pain and enlarged upper limbs. Reumatol Clin. 2011; 7(3):210–211.8. Plaiasu V, Costin A. Skeletal dysplasia presenting as a neuromuscular disorder: report of a family with Camurati-Engelmann syndrome. Maedica (Buchar). 2015; 10(1):48–51.9. Stenzler S, Grogan DP, Frenchman SM, McClelland S, Ogden JA. Progressive diaphyseal dysplasia presenting as neuromuscular disease. J Pediatr Orthop. 1989; 9(4):463–467.
Article10. Low LC, Stephenson JB, Stuart-Smith DA. Progressive diaphyseal dysplasia mimicking childhood myopathy: clinical and biochemical response to prednisolone. Aust Paediatr J. 1985; 21(3):193–196.
Article11. Sparkes RS, Graham CB. Camurati-Engelmann disease: genetics and clinical manifestations with a review of the literature. J Med Genet. 1972; 9(1):73–85.
Article12. Baretic M, Korsic M, Potocki K, Horvatic GH, Orlic ZC. Camurati-Engelmann disease in a family from Croatian Island: an old bone scan confirmed pattern of inheritance. Coll Antropol. 2014; 38(2):755–758.13. Bondestam J, Pihko H, Vanhanen SL, et al. Skeletal dysplasia presenting as a neuromuscular disorder: report of three children. Neuromuscul Disord. 2007; 17(3):231–234.
Article14. Yoshioka H, Mino M, Kiyosawa N, et al. Muscular changes in Engelmann's disease. Arch Dis Child. 1980; 55(9):716–719.
Article15. Massague J, Cheifetz S, Endo T, Nadal-Ginard B. Type beta transforming growth factor is an inhibitor of myogenic differentiation. Proc Natl Acad Sci U S A. 1986; 83(21):8206–8210.
Article16. Ignotz RA, Massague J. Type beta transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc Natl Acad Sci U S A. 1985; 82(24):8530–8534.
Article17. Simsek S, Janssens K, Kwee ML, Van Hul W, Veenstra J, Netelenbos JC. Camurati-Engelmann disease (progressive diaphyseal dysplasia) in a Moroccan family. Osteoporos Int. 2005; 16(9):1167–1170.
Article18. Collet C, Laplanche JL, de Vernejoul MC. Camurati-Engelmann disease with obesity in a newly identified family carrying a missense p.Arg156Cys mutation in the TGFB1 gene. Am J Med Genet A. 2013; 161A(8):2074–2077.19. Bhadada SK, Sridhar S, Steenackers E, et al. Camurati-Engelmann disease (progressive diaphyseal dysplasia): reports of an Indian kindred. Calcif Tissue Int. 2014; 94(2):240–247.
Article20. Girdany BR. Engelmann's disease (progressive diaphyseal dysplasia): a nonprogressive familial form of muscular dystrophy with characteristic bone changes. Clin Orthop. 1959; 14:102–109.21. Wallace SE, Wilcox WR. Camurati-Engelmann disease [Internet]. Seattle, WA: University of Washington;2015. cited 2015 Dec 31. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1156/.22. Simsek-Kiper PO, Dikoglu E, Campos-Xavier B, et al. Positive effects of an angiotensin II type 1 receptor antagonist in Camurati-Engelmann disease: a single case observation. Am J Med Genet A. 2014; 164A(10):2667–2671.
Article23. Vanhoenacker FM, Janssens K, Van Hul W, Gershoni-Baruch R, Brik R, De Schepper AM. Camurati-Engelmann disease: review of radioclinical features. Acta Radiol. 2003; 44(4):430–434.
Article24. Clybouw C, Desmyttere S, Bonduelle M, Piepsz A. Camurati-Engelmann disease: contribution of bone scintigraphy to genetic counseling. Genet Couns. 1994; 5(2):195–198.25. Castro GR, Appenzeller S, Marques-Neto JF, Bertolo MB, Samara AM, Coimbra I. Camurati-Engelmann disease: failure of response to bisphosphonates: report of two cases. Clin Rheumatol. 2005; 24(4):398–401.
Article26. Simsek-Kiper PO, Dikoglu E, Campos-Xavier B, et al. Positive effects of an angiotensin II type 1 receptor antagonist in Camurati-Engelmann disease: a single case observation. Am J Med Genet A. 2014; 164A(10):2667–2671.
Article27. Ayyavoo A, Derraik JG, Cutfield WS, Hofman PL. Elimination of pain and improvement of exercise capacity in Camurati-Engelmann disease with losartan. J Clin Endocrinol Metab. 2014; 99(11):3978–3982.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Regarding Camurati-Engelmann Disease: In Reply
- Regarding Camurati-Engelmann Disease: To the Editor
- Camurati-Engelmann's Disease on (99m)Tc-MDP Bone Scan
- The First Korean Case of Camurati-Engelmann Disease (Progressive Diaphyseal Dysplasia) Confirmed by TGFB1 Gene Mutation Analysis
- Progressive diaphyseal dysplasia (Engelmann's disease): report of a case
