A Mild Form of COG5 Defect Showing Early-Childhood-Onset Friedreich's-Ataxia-Like Phenotypes with Isolated Cerebellar Atrophy
- Affiliations
-
- 1Department of Pediatrics, Chonnam National University Medical School, Gwangju, Korea. ik052@jnu.ac.kr
- 2Department of Microbiology, Chonnam National University Medical School, Gwangju, Korea.
- 3Department of Neurology, Chonnam National University Medical School, Gwangju, Korea.
- 4Department of Radiology, Chonnam National University Medical School, Gwangju, Korea.
- 5School of Biological Sciences and Technology, Chonnam National University, Gwangju, Korea.
- KMID: 2390320
- DOI: http://doi.org/10.3346/jkms.2017.32.11.1885
Abstract
- Progressive cerebellar ataxias are rare diseases during childhood, especially under 6 years of age. In a single family, three affected siblings exhibited Friedreich's-ataxia-like phenotypes before 2 years of age. They had progressive cerebellar atrophy, intellectual disability, and scoliosis. Although their phenotypes were similar to those observed in patients with autosomal recessive cerebellar ataxias, other phenotypes (e.g., seizure, movement disorders, ophthalmologic disturbance, cardiomyopathy, and cutaneous disorders) were not noted in this family. Whole-exome sequencing of the family members revealed one potential heterozygous mutation (c.1209delG, NM_181733.2; p.Met403IlefsX3, NP_859422.2) of the gene encoding conserved oligomeric Golgi complex subunit 5 (COG5). The heterozygous deletion at the fifth base in exon 12 of COG5 caused a frameshift and premature stop. Western blotting of COG5 proteins in the skin tissues from an affected proband showed a significantly decreased level of full length COG5 and smaller, aberrant COG5 proteins. We reported a milder form of COG5 defect showing Friedreich's-ataxia-like phenotypes without hypotonia, microcephaly, and short stature that were observed in most patients with COG5 defect.
Keyword
MeSH Terms
Figure
-
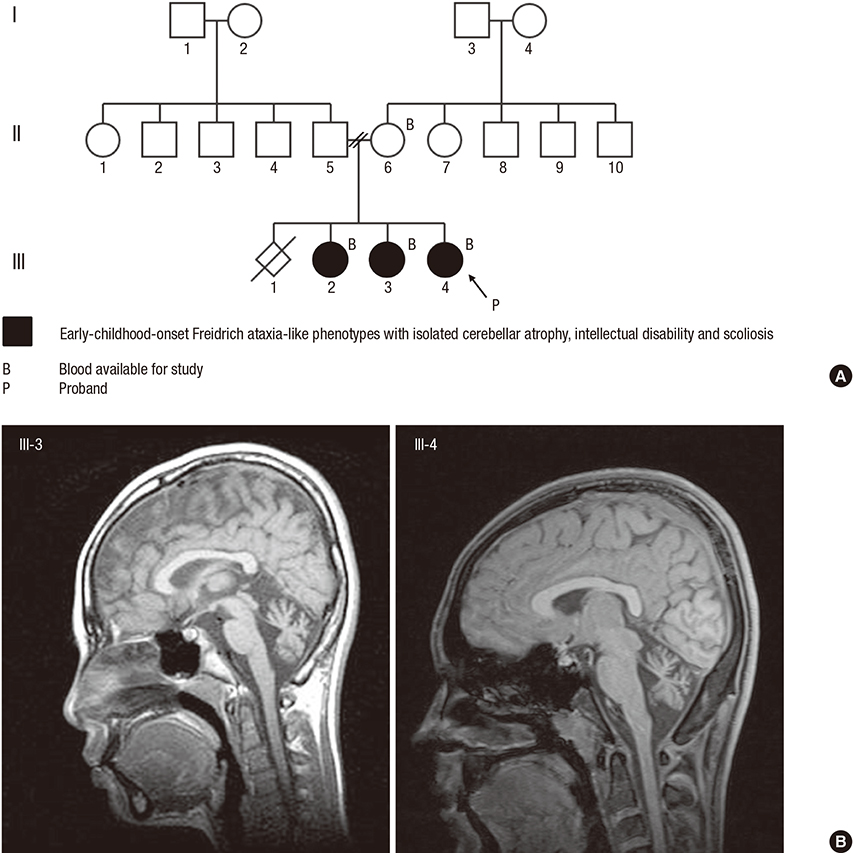
Fig. 1 Family pedigree and brain MRI. (A) Family pedigree of the family with early-childhood-onset Friedreich's-ataxia-like phenotype and isolated cerebellar atrophy. (B) Midsagittal T1-weighted brain MRI in patients III-3 and III-4 (proband) showing cerebellar atrophy with enlarged interfolial spaces in the cerebellum. No abnormalities in other parts of brain parenchyma were noted. MRI = magnetic resonance imaging.
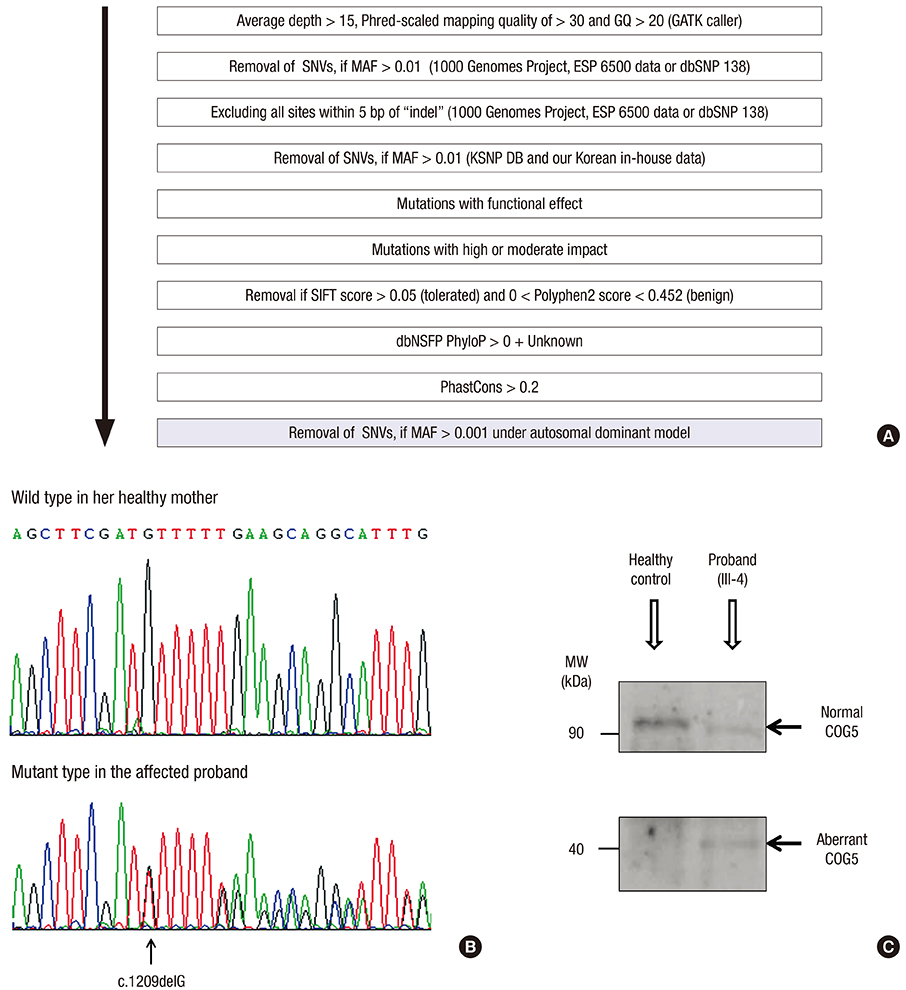
Fig. 2 Mutation analysis of the gene encoding COG5. (A) Bioinformatic prioritization for variants in WES. (B) Electropherograms showing the wild-type reference sequence of COG5 in the healthy mother, and a heterozygous deletion (arrow) at the fifth base in exon 12 of COG5 causing a frameshift and premature stop (c.1209delG, NM_181733.2; p.Met403IlefsX3, NP_859422.2) in the affected proband. (C) Western blotting of COG5 skin proteins from the affected proband and a healthy control with an anti-COG5 antibody. The proband had two bands at about 90 and 40 kDa, representing the normal and aberrant COG5 proteins, respectively. The level of full-length COG5 at about 90 kDa in the proband appears to be significantly decreased compared to that in the normal healthy control showing a single meaningful band. COG5 = conserved oligomeric Golgi complex subunit 5, WES = whole-exome sequencing, GQ = genotype quality score, GATK = Genome Analysis Toolkit, SNVs = single-nucleotide variants, MAF = minor allele frequency, ESP = Exome Sequencing Project, dbSNP = Single Nucleotide Polymorphism Database, KSNP DB = Korean Single Nucleotide Polymorphism Database, SIFT = Sorting Intolerant From Tolerant, Polyphen2 = Polymorphism Phenotyping v2, dbNSFP = Database for Nonsynonymous SNPs Functional Predictions, PhyloP = phylogenetic P value, MW = molecular weight.
Reference
-
1. Ashley CN, Hoang KD, Lynch DR, Perlman SL, Maria BL. Childhood ataxia: clinical features, pathogenesis, key unanswered questions, and future directions. J Child Neurol. 2012; 27:1095–1120.2. Fogel BL, Perlman S. Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol. 2007; 6:245–257.3. Brusse E, Maat-Kievit JA, van Swieten JC. Diagnosis and management of early- and late-onset cerebellar ataxia. Clin Genet. 2007; 71:12–24.4. Poretti A, Wolf NI, Boltshauser E. Differential diagnosis of cerebellar atrophy in childhood. Eur J Paediatr Neurol. 2008; 12:155–167.5. Zeevaert R, Foulquier F, Jaeken J, Matthijs G. Deficiencies in subunits of the Conserved Oligomeric Golgi (COG) complex define a novel group of Congenital Disorders of Glycosylation. Mol Genet Metab. 2008; 93:15–21.6. Jaeken J. Congenital disorders of glycosylation (CDG): it’s (nearly) all in it! J Inherit Metab Dis. 2011; 34:853–858.7. Wu X, Steet RA, Bohorov O, Bakker J, Newell J, Krieger M, Spaapen L, Kornfeld S, Freeze HH. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med. 2004; 10:518–523.8. Morava E, Zeevaert R, Korsch E, Huijben K, Wopereis S, Matthijs G, Keymolen K, Lefeber DJ, De Meirleir L, Wevers RA. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur J Hum Genet. 2007; 15:638–645.9. Foulquier F, Vasile E, Schollen E, Callewaert N, Raemaekers T, Quelhas D, Jaeken J, Mills P, Winchester B, Krieger M, et al. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc Natl Acad Sci USA. 2006; 103:3764–3769.10. Foulquier F, Ungar D, Reynders E, Zeevaert R, Mills P, García-Silva MT, Briones P, Winchester B, Morelle W, Krieger M, et al. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum Mol Genet. 2007; 16:717–730.11. Paesold-Burda P, Maag C, Troxler H, Foulquier F, Kleinert P, Schnabel S, Baumgartner M, Hennet T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum Mol Genet. 2009; 18:4350–4356.12. Fung CW, Matthijs G, Sturiale L, Garozzo D, Wong KY, Wong R, Wong V, Jaeken J. COG5-CDG with a mild neurohepatic presentation. JIMD Rep. 2012; 3:67–70.13. Rymen D, Keldermans L, Race V, Régal L, Deconinck N, Dionisi-Vici C, Fung CW, Sturiale L, Rosnoblet C, Foulquier F, et al. COG5-CDG: expanding the clinical spectrum. Orphanet J Rare Dis. 2012; 7:94.14. Reynders E, Foulquier F, Leão Teles E, Quelhas D, Morelle W, Rabouille C, Annaert W, Matthijs G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum Mol Genet. 2009; 18:3244–3256.15. Shaheen R, Ansari S, Alshammari MJ, Alkhalidi H, Alrukban H, Eyaid W, Alkuraya FS. A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J Med Genet. 2013; 50:431–436.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Early Onset Cerebellar Ataxia with Retained Tendon Reflexes
- Early Onset Cerebellar Ataxia with Retained Tendon Reflexes Developed in Brothers: Report of two cases
- A Case of Friedreich's Ataxia with Optic Atrophy as an Initial Clinical Manifestation
- Spinocerebellar Ataxia Type 8 Presenting as Ataxia without Definite Cerebellar Atrophy
- The Etiologies of Chronic Progressive Cerebellar Ataxia in a Korean Population
