J Clin Neurol.
2017 Jan;13(1):91-97. 10.3988/jcn.2017.13.1.91.
Duchenne Muscular Dystrophy and Becker Muscular Dystrophy Confirmed by Multiplex Ligation-Dependent Probe Amplification: Genotype-Phenotype Correlation in a Large Cohort
- Affiliations
-
- 1Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore, India. atchayaramnalini@yahoo.co.in
- 2Department of Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, India.
- 3Department of Molecular Genetics, National Institute of Mental Health and Neurosciences, Bangalore, India.
- 4Department of Psychiatric Social Work, National Institute of Mental Health and Neurosciences, Bangalore, India.
- KMID: 2364903
- DOI: http://doi.org/10.3988/jcn.2017.13.1.91
Abstract
- BACKGROUND AND PURPOSE
Studies of cases of Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) confirmed by multiplex ligation-dependent probe amplification (MLPA) have determined the clinical characteristics, genotype, and relations between the reading frame and phenotype for different countries. This is the first such study from India.
METHODS
A retrospective genotype-phenotype analysis of 317 MLPA-confirmed patients with DMD or BMD who visited the neuromuscular clinic of a quaternary referral center in southern India.
RESULTS
The 317 patients comprised 279 cases of DMD (88%), 32 of BMD (10.1%), and 6 of intermediate phenotype (1.9%). Deletions accounted for 91.8% of cases, with duplications causing the remaining 8.2%. There were 254 cases of DMD (91%) with deletions and 25 (9%) due to duplications, and 31 cases (96.8%) of BMD with deletions and 1 (3.2%) due to duplication. All six cases of intermediate type were due to deletions. The most-common mutation was a single-exon deletion. Deletions of six or fewer exons constituted 68.8% of cases. The deletion of exon 50 was the most common. The reading-frame rule held in 90% of DMD and 94% of BMD cases. A tendency toward a lower IQ and earlier wheelchair dependence was observed with distal exon deletions, though a significant correlation was not found.
CONCLUSIONS
The reading-frame rule held in 90% to 94% of children, which is consistent with reports from other parts of the world. However, testing by MLPA is a limitation, and advanced sequencing methods including analysis of the structure of mutant dystrophin is needed for more-accurate assessments of the genotype-phenotype correlation.
MeSH Terms
Figure
-
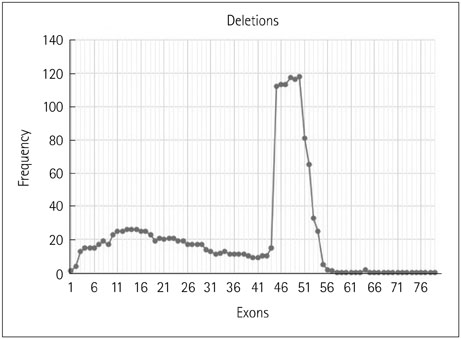
Fig. 1 Frequencies of deletions of exons in DMD and BMD. BMD: Becker muscular dystrophy, DMD: Duchenne muscular dystrophy.
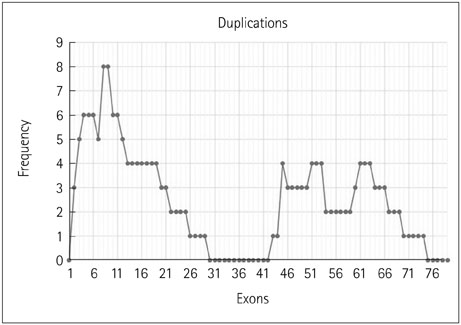
Fig. 2 Frequencies of duplications of exons in DMD and BMD. BMD: Becker muscular dystrophy, DMD: Duchenne muscular dystrophy.
Reference
-
1. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010; 9:77–93.
Article2. Bushby KM, Thambyayah M, Gardner-Medwin D. Prevalence and incidence of Becker muscular dystrophy. Lancet. 1991; 337:1022–1024.
Article3. Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature. 1986; 323:646–650.
Article4. Hoffman EP, Knudson CM, Campbell KP, Kunkel LM. Subcellular fractionation of dystrophin to the triads of skeletal muscle. Nature. 1987; 330:754–758.
Article5. Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002; 82:291–329.
Article6. Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003; 2:731–740.
Article7. Hoffman EP, Fischbeck KH, Brown RH, Johnson M, Medori R, Loike JD, et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med. 1988; 318:1363–1368.
Article8. Nicholson LV, Johnson MA, Gardner-Medwin D, Bhattacharya S, Harris JB. Heterogeneity of dystrophin expression in patients with Duchenne and Becker muscular dystrophy. Acta Neuropathol. 1990; 80:239–250.
Article9. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988; 2:90–95.
Article10. Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006; 34:135–144.
Article11. Yang J, Li SY, Li YQ, Cao JQ, Feng SW, Wang YY, et al. MLPA-based genotype-phenotype analysis in 1053 Chinese patients with DMD/BMD. BMC Med Genet. 2013; 14:29.
Article12. Luce LN, Dalamon V, Ferrer M, Parma D, Szijan I, Giliberto F, et al. MLPA analysis of an Argentine cohort of patients with dystrophinopathy: association of intron breakpoints hot spots with STR abundance in DMD gene. J Neurol Sci. 2016; 365:22–30.
Article13. Murugan S, Chandramohan A, Lakshmi BR. Use of multiplex ligation-dependent probe amplification (MLPA) for Duchenne muscular dystrophy (DMD) gene mutation analysis. Indian J Med Res. 2010; 132:303–311.14. Dey S, Senapati AK, Pandit A, Biswas A, Guin DS, Joardar A, et al. Genetic and clinical profile of patients of Duchenne muscular dystrophy: experience from a tertiary care center in Eastern India. Indian Pediatr. 2015; 52:481–484.
Article15. Swaminathan B, Shubha GN, Shubha D, Murthy AR, Kiran Kumar HB, Shylashree S, et al. Duchenne muscular dystrophy: a clinical, histopathological and genetic study at a neurology tertiary care center in Southern India. Neurol India. 2009; 57:734–738.
Article16. Rao MV, Sindhav GM, Mehta JJ. Duchenne/Becker muscular dystrophy: a report on clinical, biochemical, and genetic study in Gujarat population, India. Ann Indian Acad Neurol. 2014; 17:303–307.
Article17. Manjunath M, Kiran P, Preethish-Kumar V, Nalini A, Singh RJ, Gayathri N. A comparative study of mPCR, MLPA, and muscle biopsy results in a cohort of children with Duchenne muscular dystrophy: a first study. Neurol India. 2015; 63:58–62.
Article18. Wang X, Wang Z, Yan M, Huang S, Chen TJ, Zhong N. Similarity of DMD gene deletion and duplication in the Chinese patients compared to global populations. Behav Brain Funct. 2008; 4:20.
Article19. Lee BL, Nam SH, Lee JH, Ki CS, Lee M, Lee J. Genetic analysis of dystrophin gene for affected male and female carriers with Duchenne/Becker muscular dystrophy in Korea. J Korean Med Sci. 2012; 27:274–280.
Article20. Hwa HL, Chang YY, Chen CH, Kao YS, Jong YJ, Chao MC, et al. Multiplex ligation-dependent probe amplification identification of deletions and duplications of the Duchenne muscular dystrophy gene in Taiwanese subjects. J Formos Med Assoc. 2007; 106:339–346.
Article21. Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009; 30:934–945.
Article22. Magri F, Govoni A, D'Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011; 258:1610–1623.
Article23. White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers AL, Lalic T, et al. Duplications in the DMD gene. Hum Mutat. 2006; 27:938–945.24. Hu XY, Ray PN, Worton RG. Mechanisms of tandem duplication in the Duchenne muscular dystrophy gene include both homologous and nonhomologous intrachromosomal recombination. EMBO J. 1991; 10:2471–2477.
Article25. Zhang Z, Takeshima Y, Awano H, Nishiyama A, Okizuka Y, Yagi M, et al. Tandem duplications of two separate fragments of the dystrophin gene in a patient with Duchenne muscular dystrophy. J Hum Genet. 2008; 53:215–219.
Article26. Hattori N, Kaido M, Nishigaki T, Inui K, Fujimura H, Nishimura T, et al. Undetectable dystrophin can still result in a relatively benign phenotype of dystrophinopathy. Neuromuscul Disord. 1999; 9:220–226.
Article27. Morrone A, Zammarchi E, Scacheri PC, Donati MA, Hoop RC, Servidei S, et al. Asymptomatic dystrophinopathy. Am J Med Genet. 1997; 69:261–267.
Article28. Winnard AV, Mendell JR, Prior TW, Florence J, Burghes AH. Frameshift deletions of exons 3-7 and revertant fibers in Duchenne muscular dystrophy: mechanisms of dystrophin production. Am J Hum Genet. 1995; 56:158–166.29. Gangopadhyay SB, Sherratt TG, Heckmatt JZ, Dubowitz V, Miller G, Shokeir M, et al. Dystrophin in frameshift deletion patients with Becker muscular dystrophy. Am J Hum Genet. 1992; 51:562–570.30. Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989; 45:498–506.31. Nicolas A, Lucchetti-Miganeh C, Yaou RB, Kaplan JC, Chelly J, Leturcq F, et al. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J Rare Dis. 2012; 7:45.
Article32. Nicolas A, Raguénès-Nicol C, Ben Yaou R, Ameziane-Le Hir S, Chéron A, Vié V, et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet. 2015; 24:1267–1279.
Article33. Finsterer J, Stöllberger C. The heart in human dystrophinopathies. Cardiology. 2003; 99:1–19.
Article34. Nigro G, Politano L, Nigro V, Petretta VR, Comi LI. Mutation of dystrophin gene and cardiomyopathy. Neuromuscul Disord. 1994; 4:371–379.
Article35. Bastaki LA, Haider MZ, Shawky RM, Naguib KK. Genotype phenotype correlation among patients with dystrophinopathies. Alex J Pediatr. 1999; 13:365–371.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Female Carriers of Duchenne Muscular Dystrophy
- Novel Non-contiguous Duplications in the DMD Gene in Five Patients with Duchenne Muscular Dystrophy
- Pathologically and Genetically Diagnosed Subclinical Symptomatic Duchenne Muscular Dystrophy Carrier: Broadened Spectrum of Clinical Phenotype
- The Clinical Features, Immunostaining and Genetic Study in Duchenne/Becker Muscular Dystrophy
- Evaluation of Multiplex PCR Assay Using Dual Priming Oligonucleotide System for Detection Mutation in the Duchenne Muscular Dystrophy Gene
