Pediatr Gastroenterol Hepatol Nutr.
2014 Dec;17(4):266-269. 10.5223/pghn.2014.17.4.266.
Coexistence of Gilbert Syndrome and Hereditary Spherocytosis in a Child Presenting with Extreme Jaundice
- Affiliations
-
- 1Department of Pediatrics, Chosun University School of Medicine, Gwangju, Korea. krmoon@chosun.ac.kr
- KMID: 2315504
- DOI: http://doi.org/10.5223/pghn.2014.17.4.266
Abstract
- Gilbert syndrome is the most common inherited disorder of bilirubin glucuronidation. It is characterized by intermittent episodes of jaundice in the absence of hepatocellular disease or hemolysis. Hereditary spherocytosis is the most common inherited hemolytic anemia and is characterized by spherical, osmotically fragile erythrocytes that are selectively trapped by the spleen. The patients have variable degrees of anemia, jaundice, and splenomegaly. Hereditary spherocytosis usually leads to mild-to-moderate elevation of serum bilirubin levels. Severe hyperbilirubinemia compared with the degree of hemolysis should be lead to suspicion of additional clinical conditions such as Gilbert syndrome or thalassemia. We present the case of a 12-year-old boy with extreme jaundice and nausea. The diagnosis of hereditary spherocytosis was confirmed by osmotic fragility test results and that of Gilbert syndrome by genetic analysis findings.
Keyword
MeSH Terms
Figure
-
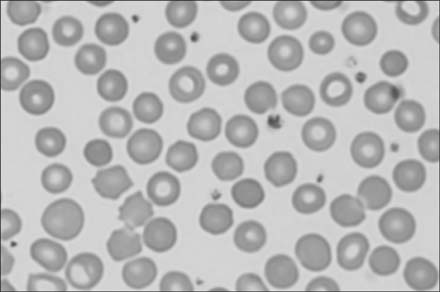
Fig. 1 Peripheral blood smear showed small and dense spherocytes (Wright & Giemsa stain, ×400).
Reference
-
1. Owens D, Evans J. Population studies on Gilbert's syndrome. J Med Genet. 1975; 12:152–156.
Article2. Fretzayas A, Moustaki M, Liapi O, Karpathios T. Gilbert syndrome. Eur J Pediatr. 2012; 171:11–15.3. Bolton-Maggs PH, Langer JC, Iolascon A, Tittensor P, King MJ. General Haematology Task Force of the British Committee for Standards in Haematology. Guidelines for the diagnosis and management of hereditary spherocytosis--2011 update. Br J Haematol. 2012; 156:37–49.
Article4. Tamary H, Aviner S, Freud E, Miskin H, Krasnov T, Schwarz M, et al. High incidence of early cholelithiasis detected by ultrasonography in children and young adults with hereditary spherocytosis. J Pediatr Hematol Oncol. 2003; 25:952–954.
Article5. Sieg A, Arab L, Schlierf G, Stiehl A, Kommerell B. Prevalence of Gilbert's syndrome in Germany. Dtsch Med Wochenschr. 1987; 112:1206–1208.6. Kim YH, Yeon JE, Jung GM, Kim HJ, Kim JS, Byun KS, et al. A study of polymorphism in UDP-glucuronosyltransferase 1 (UGT-1A1) promoter gene in Korean patients with Gilbert's syndrome. Taehan Kan Hakhoe Chi. 2002; 8:132–138.7. Muraca M, Fevery J. Influence of sex and sex steroids on bilirubin uridine diphosphate-glucuronosyltransferase activity of rat liver. Gastroenterology. 1984; 87:308–313.
Article8. Monaghan G, Ryan M, Seddon R, Hume R, Burchell B. Genetic variation in bilirubin UPD-glucuronosyltransferase gene promoter and Gilbert's syndrome. Lancet. 1996; 347:578–581.
Article9. Maruo Y, D'Addario C, Mori A, Iwai M, Takahashi H, Sato H, et al. Two linked polymorphic mutations (A(TA)7TAA and T-3279G) of UGT1A1 as the principal cause of Gilbert syndrome. Hum Genet. 2004; 115:525–526.
Article10. Yamamoto K, Sato H, Fujiyama Y, Doida Y, Bamba T. Contribution of two missense mutations (G71R and Y486D) of the bilirubin UDP glycosyltransferase (UGT1A1) gene to phenotypes of Gilbert's syndrome and Crigler-Najjar syndrome type II. Biochim Biophys Acta. 1998; 1406:267–273.
Article11. Lim JW, Choi JH, Nam YH, Seo IS, Yoon SM, Koo MS. A case of congenital hemolytic anemia of unknown cause combined with Gilbert's syndrome. Korean J Hematol. 2008; 43:58–61.
Article12. Lee MJ, Chang YH, Kang SH, Mun SK, Kim H, Han CJ, et al. A case of hereditary spherocytosis coexisting with Gilbert's syndrome. Korean J Gastroenterol. 2013; 61:166–169.
Article13. Hong YS, Jin JY, Lee WR. A case of Gilbert's syndrome with severe neonatal hyperbilirubinemia. J Korean Soc Neonatol. 2010; 17:266–269.
Article14. Lee HJ, Moon HS, Lee ES, Kim SH, Sung JK, Lee BS, et al. A case of concomitant Gilbert's syndrome and hereditary spherocytosis. Korean J Hepatol. 2010; 16:321–324.
Article15. Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008; 372:1411–1426.
Article16. Bader-Meunier B, Gauthier F, Archambaud F, Cynober T, Miélot F, Dommergues JP, et al. Long-term evaluation of the beneficial effect of subtotal splenectomy for management of hereditary spherocytosis. Blood. 2001; 97:399–403.
Article17. Iijima S, Ohzeki T, Maruo Y. Hereditary spherocytosis coexisting with UDP-glucuronosyltransferase deficiency highly suggestive of Crigler-Najjar syndrome type II. Yonsei Med J. 2011; 52:369–372.
Article18. Sharma S, Vukelja SJ, Kadakia S. Gilbert's syndrome co-existing with and masking hereditary spherocytosis. Ann Hematol. 1997; 74:287–289.
Article19. Garg PK, Kumar A, Teckchandani N, Hadke NS. Hereditary spherocytosis coexisting with Gilbert's syndrome: a diagnostic dilemma. Singapore Med J. 2008; 49:e308–e309.20. del Giudice EM, Perrotta S, Nobili B, Specchia C, d'Urzo G, Iolascon A. Coinheritance of Gilbert syndrome increases the risk for developing gallstones in patients with hereditary spherocytosis. Blood. 1999; 94:2259–2262.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Hereditary Spherocytosis Coexisting with Gilbert's Syndrome
- A case of concomitant Gilbert's syndrome and hereditary spherocytosis
- Hereditary Spherocytosis Coexisting with UDP-Glucuronosyltransferase Deficiency Highly Suggestive of Crigler-Najjar Syndrome Type II
- Prolonged Extreme Thrombocytosis in a Postsplenectomy Patient with Hereditary Spherocytosis
- A Case of Moyamoya Syndrome Associated with Hereditary Spherocytosis
