Genetic Variation of SCN5A in Korean Patients with Sick Sinus Syndrome
- Affiliations
-
- 1Division of Cardiology, Catholic University of Daegu, Daegu, Korea.
- 2Krannert Institute of Cardiology, Indiana University School of Medicine, Indianapolis, IN, USA.
- 3Daegu Fatima Hospital, Daegu, Korea.
- 4Yeungnam University, Daegu, Korea.
- 5Department of Internal Medicine, Keimyung University, Daegu, Korea. ynkim@dsmc.or.kr
- 6Kyungpook National University, Daegu, Korea.
- 7D&P Biotech, Daegu, Korea.
- 8Department of Biochemistry and Cell Biology, Kyungpook National University, Daegu, Korea.
- KMID: 2223771
- DOI: http://doi.org/10.4070/kcj.2016.46.1.63
Abstract
- BACKGROUND AND OBJECTIVES
Due to recent studies that have shown an association between the genetic variation of SCN5A and sick sinus syndrome (SSS), we sought to determine if a similar correlation existed in Korean patients with SSS.
SUBJECTS AND METHODS
We enrolled 30 patients with SSS who showed a sinus pause (longer than 3.0 s) in Holter monitoring, in addition to 80 controls. All exons including the putative splicing sites of the SCN5A gene were amplified by polymerase chain reaction and sequenced either directly or following subcloning. Wild-type and single nucleotide polymorphisms were expressed in human embryonic kidney cells, and the peak sodium current (I(Na)) was analyzed using the whole-cell patch-clamp technique.
RESULTS
A total of 9 genetic variations were identified: 7 variations (G87A-A29A, IVS9-3C>A, A1673G-H558R, G3823A-D1275N, T5457C-D1819D, T5963G-L1988R, and C5129T-S1710L) had been previously reported, and 2 variants (A3075T-E1025D and T4847A-F1616Y) were novel; the potential structural effects of F1616Y were analyzed in a three-dimensional model of the SCN5A domain. Patch-clamp studies at room temperature demonstrated that the peak I(Na) was significantly increased by 140% in HEK cells transfected with F1616Y compared with wild-type (-335.13 pA/pF+/-24.04, n=8 vs. -139.95 pA/pF+/-23.76, n=7, respectively). Furthermore, the voltage dependency of the activation and steady-state inactivation of F1616Y were leftward-shifted compared with wild-type (V(h) activation=-55.36 mv+/-0.22, n=8 vs. V(h) activation=-44.21 mV+/-0.17, n=7; respectively; V(h) inactivation=-104.47 mV+/-0.21, n=7 vs. V(h) inactivation=-84.89 mV+/-0.09, n=12, respectively).
CONCLUSION
F1616Y may be associated with SSS.
MeSH Terms
Figure
-

Fig. 1 DNA sequencing analysis results of the newly discovered SCN5A gene variants in this study. (A) DNA sequencing results of SCN5A exon 17. A3075T is a heterozygous nucleotide change, and causes an amino acid change from glutamine to aspartate. (B) DNA sequencing results of SCN5A exon 28. T4847A is a heterozygous nucleotide change, and causes an amino acid change from phenylalanine to tyrosine. All DNA sequence electropherograms show non-synonymous nucleotide changes in the sick sinus syndrome patients.

Fig. 2 Two variation sites (F1616Y and S1710L) within the 3D model of the SCN5A domain. F1616Y is located in the center of the second loop, which is well exposed, while the S1710L variation is located immediately following the 6th alpha helix.
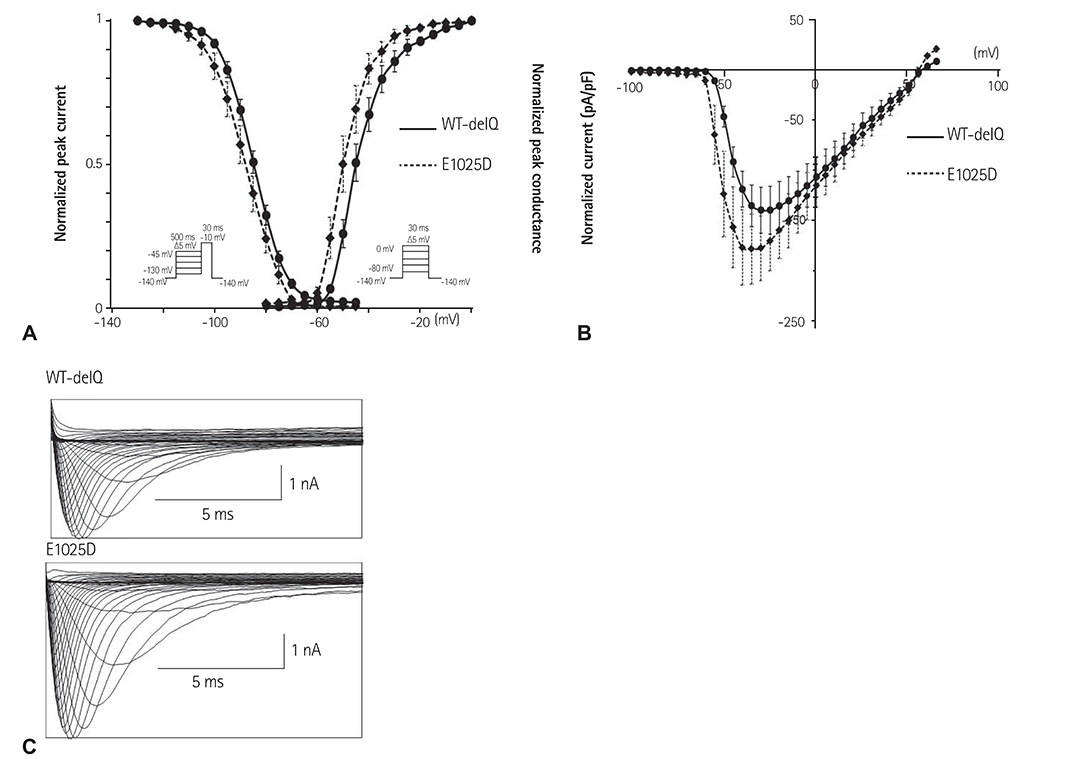
Fig. 3 Gating kinetics of WT and mutant E1025D sodium channels at 25℃ in HEK293 cells. (A) Voltage-dependent kinetics of the steady-state fast inactivation and peak conductance at 25℃. Steady-state inactivation was estimated by 500-ms pre-pulse protocols from a holding potential of -140 mV. Normalized peak currents were plotted against the pre-pulse membrane potentials. Conductance G (V) was calculated by the equation: G (V)=Ipeak/(Vm-Erev), where Ipeak is the peak current, Erev is the measured reversal potential, and Vm is the membrane potential. The normalized peak conductance was plotted against the membrane potentials. Inactivation data were fitted with the Boltzmann equation: y={1+exp ([Vm-Vh]/k)}-1; conductance data were fitted with the Boltzmann equation: y={1+ exp ([Vh-Vm]/k)}-1, where y represents variables; Vh, midpoint; k, slope factor; and Vm, membrane potential. Note: Vh (E1025D-delQ)=-90.29 mv±0.31 (n=12) is slightly leftward-shifted compared with Vh (WT-delQ)=-84.89 mV±0.09 (n=10), p<0.1; k (E1025D-delQ)=7.73±0.27, k (WT-delQ)=6.33±0.08. Vh activation (E1025D-delQ)=-52.14 mv±0.43 (n=6) is significantly leftward-shifted compared with Vh activation (WT-delQ)=-44.21 mV±0.17 (n=7) (p<0.05). Data are presented as the mean±SE. (B) I-V relationships at 25℃. Ipeak (E1025D-delQ)=-178.61 pA/pF±34.75 (n=6), Ipeak (WT-delQ)=-139.95 pA/pF±23.76 (n=7) (p<0.1). (C) Superimposed macroscopic sodium current traces obtained from the cells expressing WT-delQ (upper panel) and E1025D-delQ (lower panel). The currents were induced by step-pulse protocols from a holding potential of -140 mV. WT: wild type, SE: standard error.
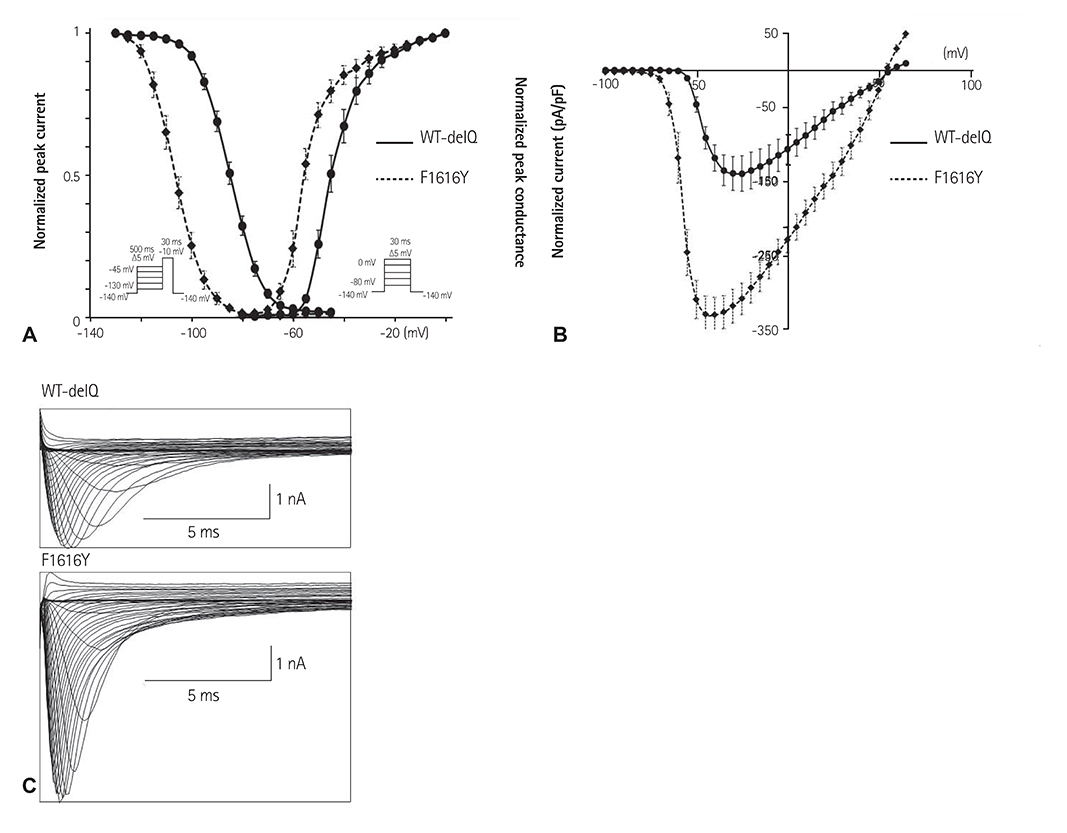
Fig. 4 Gating kinetics of WT and mutant F1616Y sodium channels at 25℃ in HEK293 cells. (A) Voltage-dependent kinetics of the steady-state fast inactivation and peak conductance at 25℃. Steady-state inactivation was estimated by 500-ms pre-pulse protocols from a holding potential of -140 mV. Normalized peak currents were plotted against the pre-pulse membrane potentials. Conductance G (V) was calculated by the equation: G (V)=Ipeak/(Vm-Erev), where Ipeak is the peak current, Erev is the measured reversal potential, and Vm is the membrane potential. The normalized peak conductance was plotted against the membrane potentials. Inactivation data were fitted with the Boltzmann equation: y={1+exp ([Vm-Vh]/k)}-1; conductance data were fitted with the Boltzmann equation: y={1+exp ([Vh-Vm]/k)}-1, where y represents variables; Vh, midpoint; k, slope factor; and Vm, membrane potential. Note: Vh (F1616Y-delQ)=-104.47 mV±0.21 (n=8) is significantly leftward-shifted compared with Vh (WT-delQ)=-84.89 mV±0.09 (n=10), p<0.005; k (F1616Y-delQ)=6.52±0.18, k (WT-delQ)=6.33±0.08. Vh activation (F1616Y-delQ)=-55.36 mv±0.22 (n=8) is significantly leftward-shifted compared with Vh activation (WT-delQ)=-44.21 mV±0.17 (n=7), p<0.005). Data are presented as the mean±SE. (B) I-V relationships at 25℃. Ipeak (F1616Y-delQ)=-335.13 pA/pF±24.04 (n=8), Ipeak (WT-delQ)=-139.95 pA/pF±23.76 (n=7) (p<0.005). (C) Superimposed macroscopic sodium current traces obtained from the cells expressing WT-delQ (upper panel) and F1616Y-delQ (lower panel). The currents were induced by step-pulse protocols from a holding potential of -140 mV. WT: wild type, SE: standard error.
Reference
-
1. Mangrum JM, DiMarco JP. The evaluation and management of bradycardia. N Engl J Med. 2000; 342:703–709.2. Benson DW, Wang DW, Dyment M, et al. Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J Clin Invest. 2003; 112:1019–1028.3. Groenewegen WA, Firouzi M, Bezzina CR, et al. A cardiac sodium channel mutation cosegregates with a rare connexin40 genotype in familial atrial standstill. Circ Res. 2003; 92:14–22.4. Lei M, Zhang H, Grace AA, Huang CL. SCN5A and sinoatrial node pacemaker function. Cardiovasc Res. 2007; 74:356–365.5. Schulze-Bahr E, Eckardt L, Breithardt G, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: Different incidences in familial and sporadic disease. Hum Mutat. 2003; 21:651–652.6. Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003; 57:961–973.7. Shin CH, Kim NH, Kim KH, et al. A family with a missense mutation in the SCN5A gene. Korean Circ J. 2003; 33:150–154.8. Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006; 22:195–201.9. Kiefer F, Arnold K, Künzli M, Bordoli L, Schwede T. The SWISS-MODEL Repository and associated resources. Nucleic Acids Res. 2009; 37(Database issue):D387–D392.10. Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003; 31:3381–3385.11. Turker I, Yu CC, Chang PC, et al. Amiodarone inhibits apamin-sensitive potassium currents. PLoS One. 2013; 8:e70450.12. Xi Y, Ai T, De Lange E, et al. Loss of function of hNav1.5 by a ZASP1 mutation associated with intraventricular conduction disturbances in left ventricular noncompaction. Circ Arrhythm Electrophysiol. 2012; 5:1017–1026.13. Gellens ME, George AL Jr, Chen LQ, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci U S A. 1992; 89:554–558.14. Lehtinen AB, Daniel KR, Shah SA, et al. Relationship between genetic variants in myocardial sodium and potassium channel genes and QT interval duration in diabetics: The Diabetes Heart Study. Ann Noninvasive Electrocardiol. 2009; 14:72–79.15. Chen JZ, Xie XD, Wang XX, Tao M, Shang YP, Guo XG. Single nucleotide polymorphisms of the SCN5A gene in Han Chinese and their relation with Brugada syndrome. Chin Med J (Engl). 2004; 117:652–656.16. Shin DJ, Jang Y, Park HY, et al. Genetic analysis of the cardiac sodium channel gene SCN5A in Koreans with Brugada syndrome. J Hum Genet. 2004; 49:573–578.17. Takahata T, Yasui-Furukori N, Sasaki S, et al. Nucleotide changes in the translated region of SCN5A from Japanese patients with Brugada syndrome and control subjects. Life Sci. 2003; 72:2391–2399.18. Gui J, Wang T, Trump D, Zimmer T, Lei M. Mutation-specific effects of polymorphism H558R in SCN5A-related sick sinus syndrome. J Cardiovasc Electrophysiol. 2010; 21:564–573.19. Gui J, Wang T, Jones RP, Trump D, Zimmer T, Lei M. Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One. 2010; 5:e10985.20. Cha K, Reeves PJ, Khorana HG. Structure and function in rhodopsin: destabilization of rhodopsin by the binding of an antibody at the N-terminal segment provides support for involvement of the latter in an intradiscal tertiary structure. Proc Natl Acad Sci U S A. 2000; 97:3016–3021.21. Jurkat-Rott K, Mitrovic N, Hang C, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci U S A. 2000; 97:9549–9554.22. Makita N, Sasaki K, Groenewegen WA, et al. Congenital atrial standstill associated with coinheritance of a novel SCN5A mutation and connexin 40 polymorphisms. Heart Rhythm. 2005; 2:1128–1134.23. Baruscotti M, DiFrancesco D, Robinson RB. A TTX-sensitive inward sodium current contributes to spontaneous activity in newborn rabbit sino-atrial node cells. J Physiol. 1996; 492(Pt 1):21–30.24. Tan HL, Bink-Boelkens MT, Bezzina CR, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001; 409:1043–1047.25. Veldkamp MW, Wilders R, Baartscheer A, Zegers JG, Bezzina CR, Wilde AA. Contribution of sodium channel mutations to bradycardia and sinus node dysfunction in LQT3 families. Circ Res. 2003; 92:976–983.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- SCN5A p.P1725L variant that showed ventricular fibrillation and recurrent pericarditis, and a family member with sick sinus syndrome
- Electrical Injury As A Possible Cause of Sick Sinus Syndrome
- A Case Report of Neuroleptanesthesia in a Parturient with Sick Sinus Syndrome
- General Anesthesia with Laryngeal Mask Airway for Operation of a Patient with Sick - Sinus Syndrome
- Thyrotoxicosis Induced Sick Sinus Syndrome
