J Clin Neurol.
2013 Oct;9(4):274-279. 10.3988/jcn.2013.9.4.274.
Non-Ataxic Phenotypes of SCA8 Mimicking Amyotrophic Lateral Sclerosis and Parkinson Disease
- Affiliations
-
- 1Department of Neurology, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. jinwhan.cho@samsung.com
- 2Department of Neurology, Soonchunhyang University Hospital, Soonchunhyang University School of Medicine, Seoul, Korea.
- 3Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- KMID: 1980548
- DOI: http://doi.org/10.3988/jcn.2013.9.4.274
Abstract
- BACKGROUND
Spinocerebellar ataxia (SCA) type 8 (SCA8) is an inherited neurodegenerative disorder caused by the expansion of untranslated CTA/CTG triplet repeats on 13q21. The phenomenology of SCA8 is relatively varied when compared to the other types of SCAs and its spectrum is not well established.
CASE REPORT
Two newly detected cases of SCA8 with the nonataxic phenotype and unusual clinical manifestations such as dopaminergic-treatment-responsive parkinsonism and amyotrophic lateral sclerosis (ALS) are described herein. Family A expressed good dopaminergic treatment-responsive parkinsonism as an initial manifestation and developed mild cerebellar ataxia with additional movements, including dystonic gait and unusual oscillatory movement of the trunk, during the disease course. The proband of family B presented as probable ALS with cerebellar atrophy on brain MRI, with a positive family history (a brother with typical cerebellar ataxia) and genetic confirmation for SCA8.
CONCLUSIONS
Our findings support that the non-ataxic phenotypes could be caused by a mutation of the SCA8 locus which might affect neurons other than the cerebellum.
Keyword
MeSH Terms
Figure
-
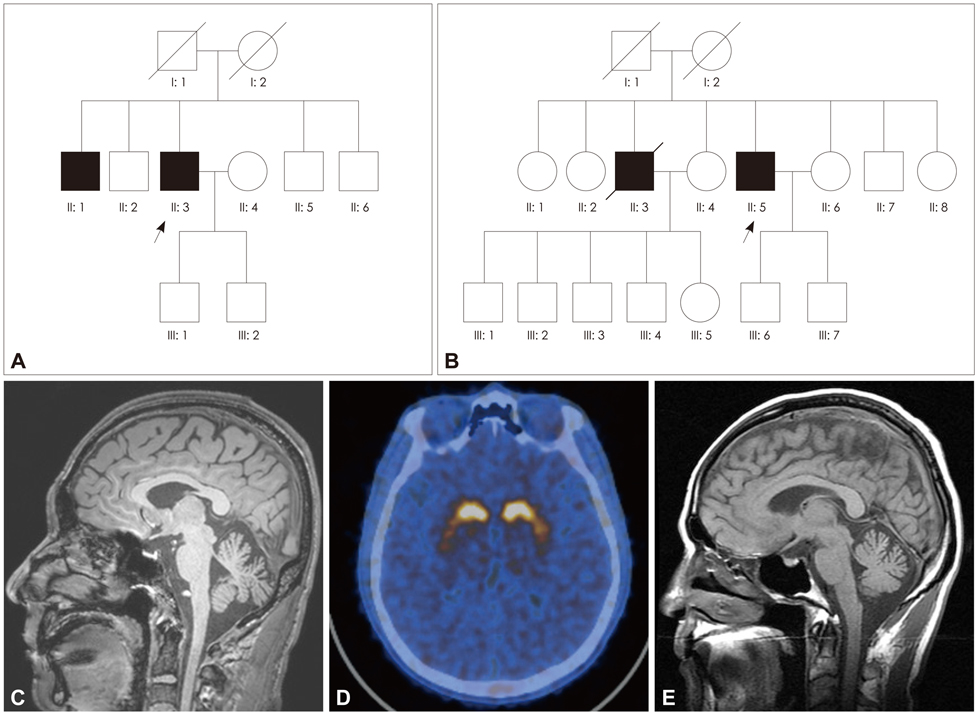
Fig. 1 A: Pedigrees of family A. B: Pedigrees of family B. Black boxes indicate the patients who express the clinical manifestations. Although II: 1 of family A revealed similar clinical phenotype with the proband (II: 3), he was not confirmed by genetic sequencing. C: Brain MRI of II: 3 (family A) revealed mild cerebellar atrophy. D: FP-CIT positron-emission tomography of II: 3 (family A) showed reduced FP-CIT binding in bilateral posterior putamen. E: Mild cerebellar atrophy was shown on brain MRI of II: 5 (family B).
Reference
-
1. Koob MD, Moseley ML, Schut LJ, Benzow KA, Bird TD, Day JW, et al. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat Genet. 1999; 21:379–384.
Article2. Day JW, Schut LJ, Moseley ML, Durand AC, Ranum LP. Spinocerebellar ataxia type 8: clinical features in a large family. Neurology. 2000; 55:649–657.
Article3. Ikeda Y, Dalton JC, Moseley ML, Gardner KL, Bird TD, Ashizawa T, et al. Spinocerebellar ataxia type 8: molecular genetic comparisons and haplotype analysis of 37 families with ataxia. Am J Hum Genet. 2004; 75:3–16.
Article4. Moseley ML, Schut LJ, Bird TD, Koob MD, Day JW, Ranum LP. SCA8 CTG repeat: en masse contractions in sperm and intergenerational sequence changes may play a role in reduced penetrance. Hum Mol Genet. 2000; 9:2125–2130.
Article5. Ikeda Y, Shizuka M, Watanabe M, Okamoto K, Shoji M. Molecular and clinical analyses of spinocerebellar ataxia type 8 in Japan. Neurology. 2000; 54:950–955.
Article6. Factor SA, Qian J, Lava NS, Hubbard JD, Payami H. False-positive SCA8 gene test in a patient with pathologically proven multiple system atrophy. Ann Neurol. 2005; 57:462–463.
Article7. Vincent JB, Yuan QP, Schalling M, Adolfsson R, Azevedo MH, Macedo A, et al. Long repeat tracts at SCA8 in major psychosis. Am J Med Genet. 2000; 96:873–876.8. Sulek A, Hoffman-Zacharska D, Zdzienicka E, Zaremba J. SCA8 repeat expansion coexists with SCA1--not only with SCA6. Am J Hum Genet. 2003; 73:972–974.
Article9. Sobrido MJ, Cholfin JA, Perlman S, Pulst SM, Geschwind DH. SCA8 repeat expansions in ataxia: a controversial association. Neurology. 2001; 57:1310–1312.
Article10. Worth PF, Houlden H, Giunti P, Davis MB, Wood NW. Large, expanded repeats in SCA8 are not confined to patients with cerebellar ataxia. Nat Genet. 2000; 24:214–215.
Article11. Juvonen V, Hietala M, Päivärinta M, Rantamäki M, Hakamies L, Kaakkola S, et al. Clinical and genetic findings in Finnish ataxia patients with the spinocerebellar ataxia 8 repeat expansion. Ann Neurol. 2000; 48:354–361.
Article12. Gupta A, Jankovic J. Spinocerebellar ataxia 8: variable phenotype and unique pathogenesis. Parkinsonism Relat Disord. 2009; 15:621–626.
Article13. Brusco A, Cagnoli C, Franco A, Dragone E, Nardacchione A, Grosso E, et al. Analysis of SCA8 and SCA12 loci in 134 Italian ataxic patients negative for SCA1-3, 6 and 7 CAG expansions. J Neurol. 2002; 249:923–929.
Article14. Tazón B, Badenas C, Jiménez L, Muñoz E, Milà M. SCA8 in the Spanish population including one homozygous patient. Clin Genet. 2002; 62:404–409.
Article15. Baba Y, Uitti RJ, Farrer MJ, Wszolek ZK. Sporadic SCA8 mutation resembling corticobasal degeneration. Parkinsonism Relat Disord. 2005; 11:147–150.
Article16. Bereznai B, Lovas G, Pentelenyi K, Rudas G, Molnar MJ. Coexisting huntingtin and SCA8 repeat expansion: case report of a severe complex neurodegenerative syndrome. J Neurol Sci. 2010; 293:116–118.
Article17. Wu YR, Chen IC, Soong BW, Kao SH, Lee GC, Huang SY, et al. SCA8 repeat expansion: large CTA/CTG repeat alleles in neurological disorders and functional implications. Hum Genet. 2009; 125:437–444.
Article18. Wu YR, Lin HY, Chen CM, Gwinn-Hardy K, Ro LS, Wang YC, et al. Genetic testing in spinocerebellar ataxia in Taiwan: expansions of trinucleotide repeats in SCA8 and SCA17 are associated with typical Parkinson's disease. Clin Genet. 2004; 65:209–214.
Article19. Nanetti L, Fancellu R, Tomasello C, Gellera C, Pareyson D, Mariotti C. Rare association of motor neuron disease and spinocerebellar ataxia type 2 (SCA2): a new case and review of the literature. J Neurol. 2009; 256:1926–1928.
Article20. Ohara S, Tsuyuzaki J, Hayashi R, Iwahashi T, Nakajima T, Maruyama T, et al. Motor neuron loss in a patient with spinocerebellar ataxia type 6: chance co-occurrence or causally related? J Neurol. 2000; 247:386–388.
Article21. Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet. 2011; 89:121–130.
Article22. Koutsis G, Karadima G, Pandraud A, Sweeney MG, Paudel R, Houlden H, et al. Genetic screening of Greek patients with Huntington's disease phenocopies identifies an SCA8 expansion. J Neurol. 2012; 259:1874–1878.
Article23. Izumi Y, Maruyama H, Oda M, Morino H, Okada T, Ito H, et al. SCA8 repeat expansion: large CTA/CTG repeat alleles are more common in ataxic patients, including those with SCA6. Am J Hum Genet. 2003; 72:704–709.
Article24. Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006; 38:758–769.
Article25. Ikeda Y, Daughters RS, Ranum LP. Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum. 2008; 7:150–158.
Article26. Chen WL, Lin JW, Huang HJ, Wang SM, Su MT, Lee-Chen GJ, et al. SCA8 mRNA expression suggests an antisense regulation of KLHL1 and correlates to SCA8 pathology. Brain Res. 2008; 1233:176–184.
Article27. Nemes JP, Benzow KA, Moseley ML, Ranum LP, Koob MD. The SCA8 transcript is an antisense RNA to a brain-specific transcript encoding a novel actin-binding protein (KLHL1). Hum Mol Genet. 2000; 9:1543–1551.
Article28. He Y, Zu T, Benzow KA, Orr HT, Clark HB, Koob MD. Targeted deletion of a single Sca8 ataxia locus allele in mice causes abnormal gait, progressive loss of motor coordination, and Purkinje cell dendritic deficits. J Neurosci. 2006; 26:9975–9982.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Neurodegenerative Disease and Speech Rehabilitation
- A Case of Frontotemporal Dementia with Amyotrophic Lateral Sclerosis Presenting with Pathological Gambling
- Syndrome of Progressive Bulbar Palsy in Amyotrophic Lateral Sclerosis: A Case Report
- Amyotrophic Lateral Sclerosis Associated With CADASIL
- Apraxia of Eyelid Closure and Motor Impersistence of Eyelid in a Patient with Amyotrophic Lateral Sclerosis
