Glucolipotoxicity in Pancreatic beta-Cells
- Affiliations
-
- 1Department of Endocrinology and Metabolism, Convergent Research Consortium for Immunologic Disease, The Catholic University of Korea, Seoul, Korea. yoonk@catholic.ac.kr
- KMID: 1857498
- DOI: http://doi.org/10.4093/dmj.2011.35.5.444
Abstract
- The recent epidemic of type 2 diabetes in Asia differs from that reported in other regions of the world in several key areas: it has evolved over a much shorter time, in an earlier stage of life, and in people with lower body mass indices. These phenotypic characteristics of patients strongly suggest that insulin secretory defects may perform a more important function in the development and progression of diabetes. A genetic element clearly underlies beta-cell dysfunction and insufficient beta-cell mass; however, a number of modifiable factors are also linked to beta-cell deterioration, most notably chronic hyperglycemia and elevated free fatty acid (FFA) levels. Neither glucose nor FFAs alone cause clinically meaningful beta-cell toxicity, especially in patients with normal or impaired glucose tolerance. Thus the term "glucolipotoxicity" is perhaps more appropriate in describing the phenomenon. Several mechanisms have been proposed to explain glucolipotoxicity-induced beta-cell dysfunction and death, but its major factors appear to be depression of key transcription factor gene expression by altered intracellular energy metabolism and oxidative stress. Therefore, stabilization of metabolic changes induced by glucolipotoxicity in beta-cells represents a new avenue for the treatment of type 2 diabetes mellitus.
MeSH Terms
Figure
-
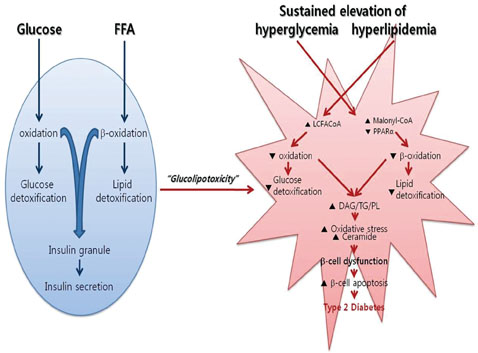
Fig. 1 Mechanism of pancreatic β-cell glucolipotoxicity. Normal laboratory ranges of glucose and fatty acids are not toxic to β-cells. The problems arise when β-cells experience prolonged exposure of sustained elevation of hyperglycemia together with elevated hyperlipidemia. Chronic high glucose levels exert a significant effect on β-cell lipid metabolism via altered enzyme activity and expression of key transcription factors. The consequent change in cellular energy metabolism results in glucolipotoxicity, which induces β-cell apoptotic cell death and promotes type 2 diabetes. FFA, free fatty acid; LCFACoA, long-chain fatty acyl-CoA; DAG, diacylglycerol; TG, triglyceride; PL, phospholipid.
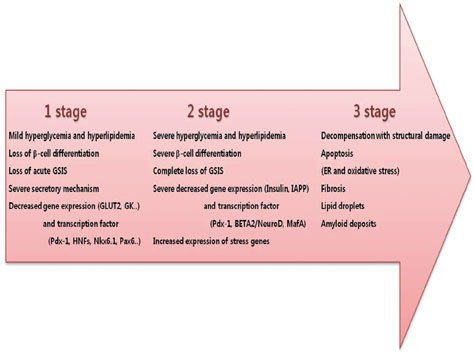
Fig. 2 Progression from the normal state to severe diabetes. This panel shows the changes that occur in β-cells during the three-stage progression from the normal state to severe diabetes. GSIS, glucose-stimulated insulin secretion; GLUT2, glucose transporter2; GK, glucokinase; IAPP, islet amyloid polypeptide; ER, endoplasmic reticulum.
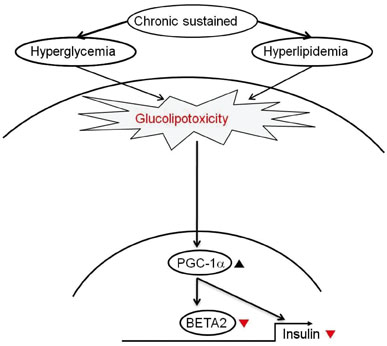
Fig. 3 Working hypothesis in glucolipotoxicity-induced β-cell dysfunction. PGC-1α inhibits insulin and BETA2/NeuroD transcription levels, and attenuating PGC-1α overexpression protects against glucolipotoxicity-induced β-cell dysfunction [13].
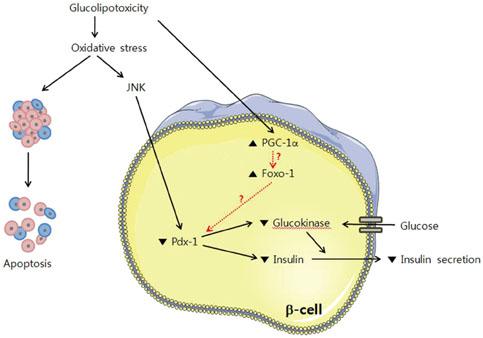
Fig. 4 β-cell dysfunction by glucolipotoxicity. Under glucolipotoxic conditions, oxidative stress is induced and the JNK pathway is activated. PDX-1 is translocated pathogenically from the nucleus to the cytoplasm by JNK activation and finally induces suppression of insulin secretion [41].
Cited by 1 articles
-
Overcoming β-Cell Dysfunction in Type 2 Diabetes Mellitus: CD36 Inhibition and Antioxidant System
Il Rae Park, Yong Geun Chung, Kyu Chang Won
Diabetes Metab J. 2025;49(1):1-12. doi: 10.4093/dmj.2024.0796.
Reference
-
1. Yoon KH, Lee JH, Kim JW, Cho JH, Choi YH, Ko SH, Zimmet P, Son HY. Epidemic obesity and type 2 diabetes in Asia. Lancet. 2006. 368:1681–1688.2. Kahn SE. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of type 2 diabetes. Diabetologia. 2003. 46:3–19.3. Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006. 116:1802–1812.4. Cho JH, Kim JW, Shin JA, Shin J, Yoon KH. Beta-cell mass in people with type 2 diabetes. J Diabetes Invest. 2011. 2:6–17.5. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, Donath MY. Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002. 110:851–860.6. Kim JW, Ko SH, Cho JH, Sun C, Hong OK, Lee SH, Kim JH, Lee KW, Kwon HS, Lee JM, Song KH, Son HY, Yoon KH. Loss of beta-cells with fibrotic islet destruction in type 2 diabetes mellitus. Front Biosci. 2008. 13:6022–6033.7. Kaiser N, Leibowitz G, Nesher R. Glucotoxicity and beta-cell failure in type 2 diabetes mellitus. J Pediatr Endocrinol Metab. 2003. 16:5–22.8. Jonas JC, Bensellam M, Duprez J, Elouil H, Guiot Y, Pascal SM. Glucose regulation of islet stress responses and beta-cell failure in type 2 diabetes. Diabetes Obes Metab. 2009. 11:Suppl 4. 65–81.9. Henquin JC. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes. 2004. 53:Suppl 3. S48–S58.10. Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Invest. 1993. 92:514–519.11. Poitout V, Olson LK, Robertson RP. Chronic exposure of betaTC-6 cells to supraphysiologic concentrations of glucose decreases binding of the RIPE3b1 insulin gene transcription activator. J Clin Invest. 1996. 97:1041–1046.12. Sharma A, Olson LK, Robertson RP, Stein R. The reduction of insulin gene transcription in HIT-T15 beta cells chronically exposed to high glucose concentration is associated with the loss of RIPE3b1 and STF-1 transcription factor expression. Mol Endocrinol. 1995. 9:1127–1134.13. Kim JW, You YH, Ham DS, Cho JH, Ko SH, Song KH, Son HY, Suh-Kim H, Lee IK, Yoon KH. Suppression of peroxisome proliferator-activated receptor gamma-coactivator-1alpha normalizes the glucolipotoxicity-induced decreased BETA2/NeuroD gene transcription and improved glucose tolerance in diabetic rats. Endocrinology. 2009. 150:4074–4083.14. Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004. 53:Suppl 3. S16–S21.15. Kajimoto Y, Kaneto H. Role of oxidative stress in pancreatic beta-cell dysfunction. Ann N Y Acad Sci. 2004. 1011:168–176.16. Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004. 279:42351–42354.17. Ritzel RA, Butler AE, Rizza RA, Veldhuis JD, Butler PC. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care. 2006. 29:717–718.18. Poitout V, Amyot J, Semache M, Zarrouki B, Hagman D, Fontes G. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta. 2010. 1801:289–298.19. Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003. 52:726–733.20. Zhou YP, Grill VE. Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest. 1994. 93:870–876.21. Tan CP, Feng Y, Zhou YP, Eiermann GJ, Petrov A, Zhou C, Lin S, Salituro G, Meinke P, Mosley R, Akiyama TE, Einstein M, Kumar S, Berger JP, Mills SG, Thornberry NA, Yang L, Howard AD. Selective small-molecule agonists of G protein-coupled receptor 40 promote glucose-dependent insulin secretion and reduce blood glucose in mice. Diabetes. 2008. 57:2211–2219.22. Alquier T, Peyot ML, Latour MG, Kebede M, Sorensen CM, Gesta S, Ronald Kahn C, Smith RD, Jetton TL, Metz TO, Prentki M, Poitout V. Deletion of GPR40 impairs glucose-induced insulin secretion in vivo in mice without affecting intracellular fuel metabolism in islets. Diabetes. 2009. 58:2607–2615.23. Nolan CJ, Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol Metab. 2008. 19:285–291.24. Boden G, Chen X, Rosner J, Barton M. Effects of a 48-h fat infusion on insulin secretion and glucose utilization. Diabetes. 1995. 44:1239–1242.25. Cnop M, Hannaert JC, Grupping AY, Pipeleers DG. Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification. Endocrinology. 2002. 143:3449–3453.26. Brunham LR, Kruit JK, Pape TD, Timmins JM, Reuwer AQ, Vasanji Z, Marsh BJ, Rodrigues B, Johnson JD, Parks JS, Verchere CB, Hayden MR. Beta-cell ABCA1 influences insulin secretion, glucose homeostasis and response to thiazolidinedione treatment. Nat Med. 2007. 13:340–347.27. Ogihara T, Chuang JC, Vestermark GL, Garmey JC, Ketchum RJ, Huang X, Brayman KL, Thorner MO, Repa JJ, Mirmira RG, Evans-Molina C. Liver X receptor agonists augment human islet function through activation of anaplerotic pathways and glycerolipid/free fatty acid cycling. J Biol Chem. 2010. 285:5392–5404.28. Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE. Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem. 1992. 267:5802–5810.29. Hagman DK, Hays LB, Parazzoli SD, Poitout V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J Biol Chem. 2005. 280:32413–32418.30. Solinas G, Naugler W, Galimi F, Lee MS, Karin M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc Natl Acad Sci U S A. 2006. 103:16454–16459.31. Joseph JW, Koshkin V, Zhang CY, Wang J, Lowell BB, Chan CB, Wheeler MB. Uncoupling protein 2 knockout mice have enhanced insulin secretory capacity after a high-fat diet. Diabetes. 2002. 51:3211–3219.32. Joseph JW, Koshkin V, Saleh MC, Sivitz WI, Zhang CY, Lowell BB, Chan CB, Wheeler MB. Free fatty acid-induced beta-cell defects are dependent on uncoupling protein 2 expression. J Biol Chem. 2004. 279:51049–51056.33. Piro S, Anello M, Di Pietro C, Lizzio MN, Patane G, Rabuazzo AM, Vigneri R, Purrello M, Purrello F. Chronic exposure to free fatty acids or high glucose induces apoptosis in rat pancreatic islets: possible role of oxidative stress. Metabolism. 2002. 51:1340–1347.34. Laybutt DR, Preston AM, Akerfeldt MC, Kench JG, Busch AK, Biankin AV, Biden TJ. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007. 50:752–763.35. Roche E, Assimacopoulos-Jeannet F, Witters LA, Perruchoud B, Yaney G, Corkey B, Asfari M, Prentki M. Induction by glucose of genes coding for glycolytic enzymes in a pancreatic beta-cell line (INS-1). J Biol Chem. 1997. 272:3091–3098.36. Roche E, Farfari S, Witters LA, Assimacopoulos-Jeannet F, Thumelin S, Brun T, Corkey BE, Saha AK, Prentki M. Longterm exposure of beta-INS cells to high glucose concentrations increases anaplerosis, lipogenesis, and lipogenic gene expression. Diabetes. 1998. 47:1086–1094.37. Farfari S, Schulz V, Corkey B, Prentki M. Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes. 2000. 49:718–726.38. Prentki M, Joly E, El-Assaad W, Roduit R. Malonyl-CoA signaling, lipid partitioning, and glucolipotoxicity: role in beta-cell adaptation and failure in the etiology of diabetes. Diabetes. 2002. 51:Suppl 3. S405–S413.39. Weir GC, Laybutt DR, Kaneto H, Bonner-Weir S, Sharma A. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes. 2001. 50:Suppl 1. S154–S159.40. Grill V, Bjorklund A. Dysfunctional insulin secretion in type 2 diabetes: role of metabolic abnormalities. Cell Mol Life Sci. 2000. 57:429–440.41. Kaneto H, Matsuoka TA, Nakatani Y, Kawamori D, Miyatsuka T, Matsuhisa M, Yamasaki Y. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med (Berl). 2005. 83:429–439.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- AICAR Reversed the Glucolipotoxicity Induced beta-cell Dysfunction through Suppression of PPAR-gamma-coactivator-1 (PGC-1) Overexpression
- The Effects of Exendin-4 on IRS-2 Expression and Phosphorylation in INS-1 Cells
- The Role of CD36 in Type 2 Diabetes Mellitus: β-Cell Dysfunction and Beyond
- Cell Replacement and Regeneration Therapy for Diabetes
- Cause of Intracellular ATP dependency on Zn2++ Blockade of KATP Channels in Pancreatic Beta Cells
