Cystic Fibrosis in Korean Children: A Case Report Identified by a Quantitative Pilocarpine Iontophoresis Sweat Test and Genetic Analysis
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. silee@smc.samsung.co.kr
- 2Department of Pharmacology, Yonsei University College of Medicine, Seoul, Korea.
- 3Department of Laboratory Medicine, Yonsei University College of Medicine, Seoul, Korea.
- 4Department of Pediatrics, Fatima Hospital, Daegu, Korea.
- KMID: 1781745
- DOI: http://doi.org/10.3346/jkms.2005.20.1.153
Abstract
- Cystic fibrosis (CF) is inherited as an autosomal recessive trait, and the mutations in cystic fibrosis transmembrane conductance regulator (CFTR) gene contributes to the CF syndrome. Although CF is common in Caucasians, it is known to be rare in Asians. Recently, we experienced two cases of CF in Korean children. The patients were girls with chronic productive cough since early infancy. Chest computed tomography showed the diffuse bronchiectasis in both lungs, and their diagnosis was confirmed by the repeated analysis of a quantitative pilocarpine iontophoresis test (QPIT). The sweat chloride concentrations of the first patient were 108.1 mM/L and 96.7 mM/L. The genetic analysis revealed that she was the compound heterozygote of Q1291X and IVS8 T5 -M470V. In the second case, the sweat chloride concentrations were 95.0 mM/L and 77.5 mM/L. Although we performed a comprehensive search for the coding regions and exonintron splicing junctions of CFTR gene, no obvious disease-related mutations were detected in the second case. To our knowledge, this is the first report of CF in Korean children identified by a QPIT and genetic analysis. The possibility of CF should be suspected in those patients with chronic respiratory symptoms even in Korea.
MeSH Terms
-
Blood Pressure
Bronchiectasis/diagnosis/pathology
Child
Cough
Cystic Fibrosis/*diagnosis/*genetics
Cystic Fibrosis Transmembrane Conductance Regulator/genetics
DNA Mutational Analysis
Exons
Female
Heterozygote
Humans
Introns
Iontophoresis/*methods
Korea
Lung/pathology/radiography
Muscarinic Agonists/*pharmacology
Mutation
Pancreas/pathology
Pedigree
Phenotype
Pilocarpine/*pharmacology
Polymorphism, Genetic
Research Support, Non-U.S. Gov't
Sinusitis/diagnosis/pathology
Sweat
Time Factors
Tomography, X-Ray Computed
Figure
-
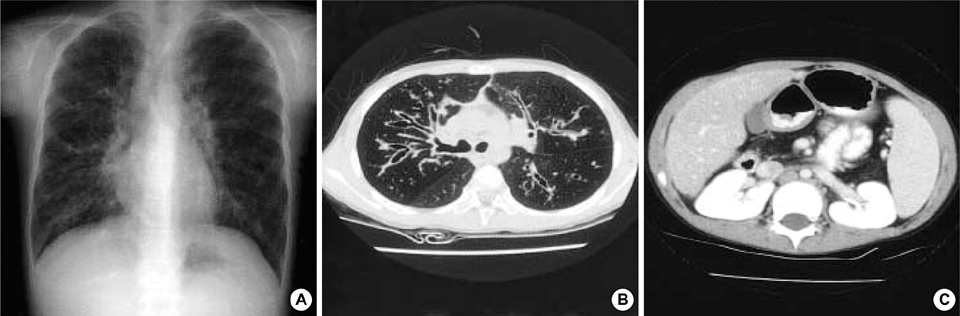
Fig. 1 Radiologic findings of case 1. Plain radiography (A) and computed tomography (CT) scan (B) reveal bronchiectasis. Pancreatic atrophy is found in abdomen CT scan (C).
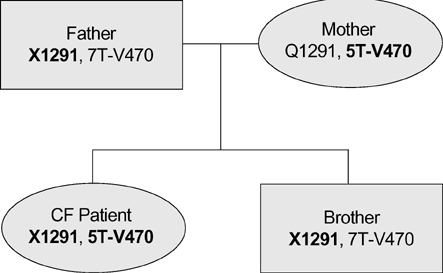
Fig. 2 Family pedigree of the disease-associated CFTR mutations in case 1. Disease associated genetic variations are shown in bold letters.
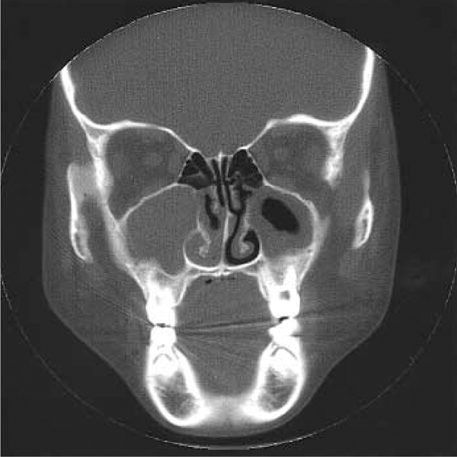
Fig. 3 CT findings, coronal view in case 2. Pansinusitis with polyposis is shown.
Cited by 7 articles
-
Lack of Association between Matrix Metalloproteinase 8 Promoter Polymorphism and Bronchiectasis in Koreans
Jiwon Lee, Hye-Ryoun Kim, Joo-Won Min, Jong Sun Park, Sang-Man Jin, Sung Koo Han, Young-Soo Shim, Jae-Joon Yim
J Korean Med Sci. 2007;22(4):667-671. doi: 10.3346/jkms.2007.22.4.667.Identification of a Novel Mutation of CFTR Gene in a Korean Patient with Cystic Fibrosis
Jung Min Ko, Gu-Hwan Kim, Kyung Mo Kim, Soo-Jong Hong, Han-Wook Yoo
J Korean Med Sci. 2008;23(5):912-915. doi: 10.3346/jkms.2008.23.5.912.Novel CFTR Mutations in a Korean Infant with Cystic Fibrosis and Pancreatic Insufficiency
Young June Choe, Jae Sung Ko, Jeong Kee Seo, Jae Jun Han, Jung Ok Shim, Young Yull Koh, Ran Lee, Chang-Seok Ki, Jong-Won Kim, Jung Ho Kim
J Korean Med Sci. 2010;25(1):163-165. doi: 10.3346/jkms.2010.25.1.163.Cystic fibrosis lung disease: Current perspectives
Jin-A Jung
Allergy Asthma Respir Dis. 2020;8(1):3-8. doi: 10.4168/aard.2020.8.1.3.Heterogeneous Spectrum of
CFTR Gene Mutations in Korean Patients with Cystic Fibrosis
Haiyoung Jung, Chang-Seok Ki, Won-Jung Koh, Kang-Mo Ahn, Sang-Il Lee, Jeong-Ho Kim, Jae Sung Ko, Jeong Kee Seo, Seung-Ick Cha, Eun-Sil Lee, Jong-Won Kim
Korean J Lab Med. 2011;31(3):219-224. doi: 10.3343/kjlm.2011.31.3.219.Literature review and future strategies of childhood respiratory diseases in Korea
Man Yong Han, Hai Lee Chung, Young Min Ahn, Jung Yeon Shim
Allergy Asthma Respir Dis. 2018;6(Suppl 1):S66-S76. doi: 10.4168/aard.2018.6.S1.S66.Standardized Sweat Chloride Analysis for the Diagnosis of Cystic Fibrosis in Korea
Sue Jung Kim, Mingoo Lee, Seung-Ick Cha, Hwa Young Park, Kang Mo Ahn, Chang-Seok Ki, Jeong-Ho Kim
Korean J Lab Med. 2008;28(4):274-281. doi: 10.3343/kjlm.2008.28.4.274.
Reference
-
1. Cystic Fibrosis Mutation Database. The Cystic Fibrosis Genetic Analysis Consortium. http://www.genet.sickkids.on.ca/cftr.2. Boat TF. Behrman RE, Kliegman RM, Jenson HB, editors. Cystic fibrosis. Nelson Textbook of Pediatrics. 2004. 17th ed. Philadelphia: Saunders;1437–1450.3. Farrell PM. Improving the health of patients with cystic fibrosis through newborn screening. Adv Pediatr. 2000. 47:79–115.4. Wright SW, Morton NE. The incidence of cystic fibrosis in Hawaii. Hawaii Med J. 1968. 27:229–232.5. Yamashiro Y, Shimizu T, Oguchi S, Shioya T, Nagata S, Ohtsuka Y. The estimated incidence of cystic fibrosis in Japan. J Pediatr Gastroenterol Nutr. 1997. 24:544–547.
Article6. Moon HR, Ko TS, Ko YY, Choi JH, Kim YC. Cystic fibrosis: A case presented with recurrent bronchiolitis in infancy in a Korean male infant. J Korean Med Sci. 1988. 3:157–162.
Article7. di Sant'Agnese PA, Daring RC, Perera GA, Shea E. Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas: Clinical significance and relationship to the disease. Pediatrics. 1953. 12:549–563.8. Schales O, Schales SS. A simple and accurate method for the determination of chloride in biological fluids. J Biol Chem. 1941. 140:879–884.
Article9. Lee JH, Choi JH, Namkung W, Hanrahan JW, Chang J, Song SY, Park SW, Kim DS, Yoon JH, Suh Y, Jang IJ, Nam JH, Kim SJ, Cho MO, Lee JE, Kim KH, Lee MG. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet. 2003. 12:2321–2332.10. Cystic Fibrosis Foundation. Patient Registry 1996 annual data report. 1997.11. di Sant'Agnese PA, Darling RC, Perera GA, Shea E. Sweat electrolyte disturbances associated with childhood pancreatic disease. Am J Med. 1953. 15:777–784.12. LeGrys VA. Sweat testing for the diagnosis of cystic fibrosis: Practical considerations. J Pediatr. 1996. 129:892–897.
Article13. Welsh MJ, Ramsey BW, Accurso F, Cutting GR. Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Cystic fibrosis. The metabolic and Molecular Bases of Inherited Disease. 2001. 8th ed. New York: McGraw Hill;5136–5139.14. LeGrys VA, Burnett RW. Current status of sweat testing in North America. Results of the College of American Pathologists needs assessment survey. Arch Pathol Lab Med. 1994. 118:865–867.15. Lemna WK, Feldman GL, Kerem BS, Fernbach SD, Zevkovich EP, O'Brien WE, Riordan JR, Collins FS, Tsui LC, Beaudet AL. Mutation analysis for heterozygote detection and the prenatal diagnosis of cystic fibrosis. N Engl J Med. 1990. 322:291–296.
Article16. Wong LJ, Alper OM, Wang BT, Lee MH, Lo SY. Two novel null mutations in a Taiwanese cystic fibrosis patient and a survey of East Asian CFTR mutations. Am J Med Genet. 2003. 120:296–298.17. Tsui LC. The spectrum of cystic fibrosis mutations. Trends Genet. 1992. 8:392–398.
Article18. Cuppens H, Lin W, Jaspers M, Costes B, Teng H, Vankeerberghen A, Jorissen M, Droogmans G, Reynaert I, Goossens M, Nilius B, Cassiman JJ. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. The polymorphic (TG)m locus explains the partial penetrance of the T5 polymorphism as a disease mutation. J Clin Invest. 1998. 101:487–496.
Article19. Feldmann D, Laroze F, Troadec C, Clement A, Tournier G, Couderc R. A novel nonsense mutation (Q1291X) in exon 20 of CFTR (ABCC7) gene. Hum Mutat. 2001. 17:356.
Article20. Groman JD, Meyer ME, Wilmott RW, Zeitlin PL, Cutting GR. Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N Engl J Med. 2002. 347:401–407.21. Stewart B, Zabner J, Shuber AP, Welsh MJ, McCray PB Jr. Normal sweat chloride values do not exclude the diagnosis of cystic fibrosis. Am J Respir Crit Care Med. 1995. 151:899–903.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Pilocarpine iontophoresis sweat test results in adults withbronchiectasis
- Standardized Sweat Chloride Analysis for the Diagnosis of Cystic Fibrosis in Korea
- Cystic fibrosis: a case presented with recurrent bronchiolitis in infancy in a Korean male infant
- Cystic Fibrosis: Case Report
- Radiologic Findings of Cystic Fibrosis in a Korean Child at Follow Up Study: Case Report
