J Vet Sci.
2009 Sep;10(3):203-210. 10.4142/jvs.2009.10.3.203.
Charting the proteome of Cryptosporidium parvum sporozoites using sequence similarity-based BLAST searching
- Affiliations
-
- 1Department of Preclinical Veterinary Sciences, Faculty of Veterinary Science, University of Liverpool, Crown Street, Liverpool, L69 7ZJ, UK.
- KMID: 1726919
- DOI: http://doi.org/10.4142/jvs.2009.10.3.203
Abstract
- Cryptosporidium (C.) spp. are important zoonotic parasites causing widespread diarrhoeal disease in man and animals. The recent release of the complete genome sequences for C. parvum and C. hominis has facilitated the comprehensive global proteome analysis of these opportunistic pathogens. The well-known approach for mass spectrometry (MS) based data analysis using the BLAST tool (MS BLAST) is a database search protocol for identifying unknown proteins by sequence similarity to homologous proteins using peptide sequences produced by mass spectrometry. We have used several complementary approaches to explore the global sporozoite proteome of C. parvum with available proteomic tools. To optimize the output of the MS data, a sequence similarity-based MS BLAST strategy was employed for bioinformatic analysis. Most significantly, almost all the constituents of glycolysis and several mitochondrion-related proteins were identified. In addition, many hypothetical Cryptosporidium proteins were validated by the identification of their constituent peptides. The MS BLAST approach was found to be useful during the study and could provide valuable information towards a complete understanding of the unique biology of Cryptosporidium.
Keyword
MeSH Terms
Figure
-
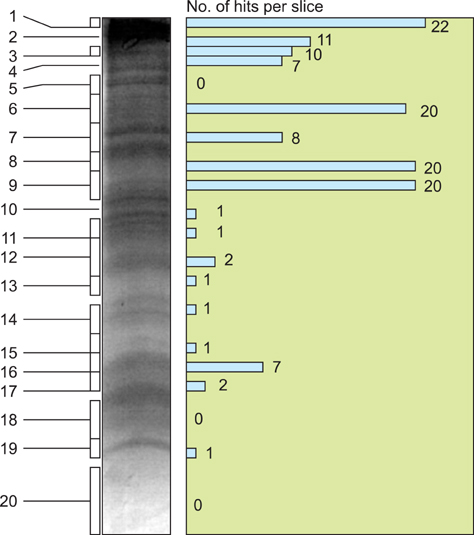
Fig. 1 Roadmap for database searches towards identifying known and putative protein sequences.
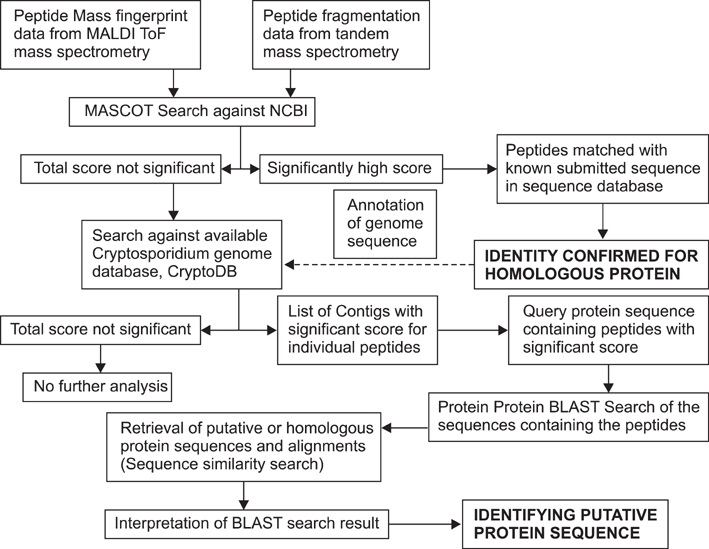
Fig. 2 First dimension SDS-PAGE of the sporozoite proteins of Cryptosporidium (C.) parvum. The lane was then excised into 20 slices and analysed by tandem mass spectrometry. The side bar shows the number of hits per slice.

Fig. 3 Functional categorization of 84 C. parvum proteins identified by mass spectrometry based BLAST searching of MS data in an 1D-SDS-PAGE experiment.
Reference
-
1. Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004. 304:441–445.
Article2. Bendtsen JD, Nielsen H, von Heijne G, Brunak S. Improved prediction of signal peptides: SignalP 3.0. J Mol Biol. 2004. 340:783–795.
Article3. Chen ZQ, Liu Q, Zhu YS, Li YX. Performance analysis of methods that predict transmembrane regions. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai). 2002. 34:285–290.4. Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002. 419:520–526.
Article5. Habermann B, Oegema J, Sunyaev S, Shevchenko A. The power and the limitations of cross-species protein identification by mass spectrometry-driven sequence similarity searches. Mol Cell Proteomics. 2004. 3:238–249.
Article6. Huang L, Jacob RJ, Pegg SC, Baldwin MA, Wang CC, Burlingame AL, Babbitt PC. Functional assignment of the 20 S proteasome from Trypanosoma brucei using mass spectrometry and new bioinformatics approaches. J Biol Chem. 2001. 276:28327–28339.
Article7. Jacoboni I, Martelli PL, Fariselli P, De Pinto V, Casadio R. Prediction of the transmembrane regions of beta-barrel membrane proteins with a neural network-based predictor. Protein Sci. 2001. 10:779–787.
Article8. Lan N, Montelione GT, Gerstein M. Ontologies for proteomics: towards a systematic definition of structure and function that scales to the genome level. Curr Opin Chem Biol. 2003. 7:44–54.
Article9. Liebel U, Kindler B, Pepperkok R. 'Harvester': a fast meta search engine of human protein resources. Bioinformatics. 2004. 20:1962–1963.
Article10. Liska AJ, Popov AV, Sunyaev S, Coughlin P, Habermann B, Shevchenko A, Bork P, Karsenti E, Shevchenko A. Homology-based functional proteomics by mass spectrometry: application to the Xenopus microtubule-associated proteome. Proteomics. 2004. 4:2707–2721.
Article11. Mackey AJ, Haystead TA, Pearson WR. Getting more from less: algorithms for rapid protein identification with multiple short peptide sequences. Mol Cell Proteomics. 2002. 1:139–147.12. Manabe YC, Clark DP, Moore RD, Lumadue JA, Dahlman HR, Belitsos PC, Chaisson RE, Sears CL. Cryptosporidiosis in patients with AIDS: correlates of disease and survival. Clin Infect Dis. 1998. 27:536–542.
Article13. Melén K, Krogh A, von Heijne G. Reliability measures for membrane protein topology prediction algorithms. J Mol Biol. 2003. 327:735–744.
Article14. Nair R, Rost B. Mimicking cellular sorting improves prediction of subcellular localization. J Mol Biol. 2005. 348:85–100.
Article15. Neuhoff V, Arold N, Taube D, Ehrhardt W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis. 1988. 9:255–262.
Article16. O'Donoghue PJ. Cryptosporidium and cryptosporidiosis in man and animals. Int J Parasitol. 1995. 25:139–195.17. Ofran Y, Punta M, Schneider R, Rost B. Beyond annotation transfer by homology: novel protein-function prediction methods to assist drug discovery. Drug Discov Today. 2005. 10:1475–1482.
Article18. Ouzounis C, Perez-Irratxeta C, Sander C, Valencia A. Are binding residues conserved? Pac Symp Biocomput. 1998. 1:401–412.19. Rost B. Enzyme function less conserved than anticipated. J Mol Biol. 2002. 318:595–608.
Article20. Sanderson SJ, Xia D, Prieto H, Yates J, Heiges M, Kissinger JC, Bromley E, Lal K, Sinden RE, Tomley F, Wastling JM. Determining the protein repertoire of Cryptosporidium parvum sporozoites. Proteomics. 2008. 8:1398–1414.
Article21. Shah I, Hunter L. Predicting enzyme function from sequence: a systematic appraisal. Proc Int Conf Intell Syst Mol Biol. 1997. 5:276–283.22. Shevchenko A, Sunyaev S, Loboda A, Shevchenko A, Bork P, Ens W, Standing KG. Charting the proteomes of organisms with unsequenced genomes by MALDI-quadrupole time-of-flight mass spectrometry and BLAST homology searching. Anal Chem. 2001. 73:1917–1926.
Article23. Todd AE, Orengo CA, Thornton JM. Evolution of function in protein superfamilies, from a structural perspective. J Mol Biol. 2001. 307:1113–1143.
Article24. Waridel P, Frank A, Thomas H, Surendranath V, Sunyaev S, Pevzner P, Shevchenko A. Sequence similarity-driven proteomics in organisms with unknown genomes by LC-MS/MS and automated de novo sequencing. Proteomics. 2007. 7:2318–2329.
Article25. Wastling JM, Xia D, Sohal A, Chaussepied M, Pain A, Langsley G. Proteomes and transcriptomes of the Apicomplexa--where's the message? Int J Parasitol. 2009. 39:135–143.26. Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q Rev Biophys. 2003. 36:307–340.
Article27. Wrzeszczynski KO, Rost B. Annotating proteins from endoplasmic reticulum and Golgi apparatus in eukaryotic proteomes. Cell Mol Life Sci. 2004. 61:1341–1353.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Sporozoite proteome analysis of Cryptosporidium parvum by one-dimensional SDS-PAGE and liquid chromatography tandem mass spectrometry
- The effect of microfilament inhibitor on the Cryptosporidium infection in vitro
- NBLAST: a graphical user interface-based two-way BLAST software with a dot plot viewer
- Ultrastructural Changes in Cryptosporidium parvum Oocysts by Gamma Irradiation
- CysQ of Cryptosporidium parvum, a Protozoa, May Have Been Acquired from Bacteria by Horizontal Gene Transfer
