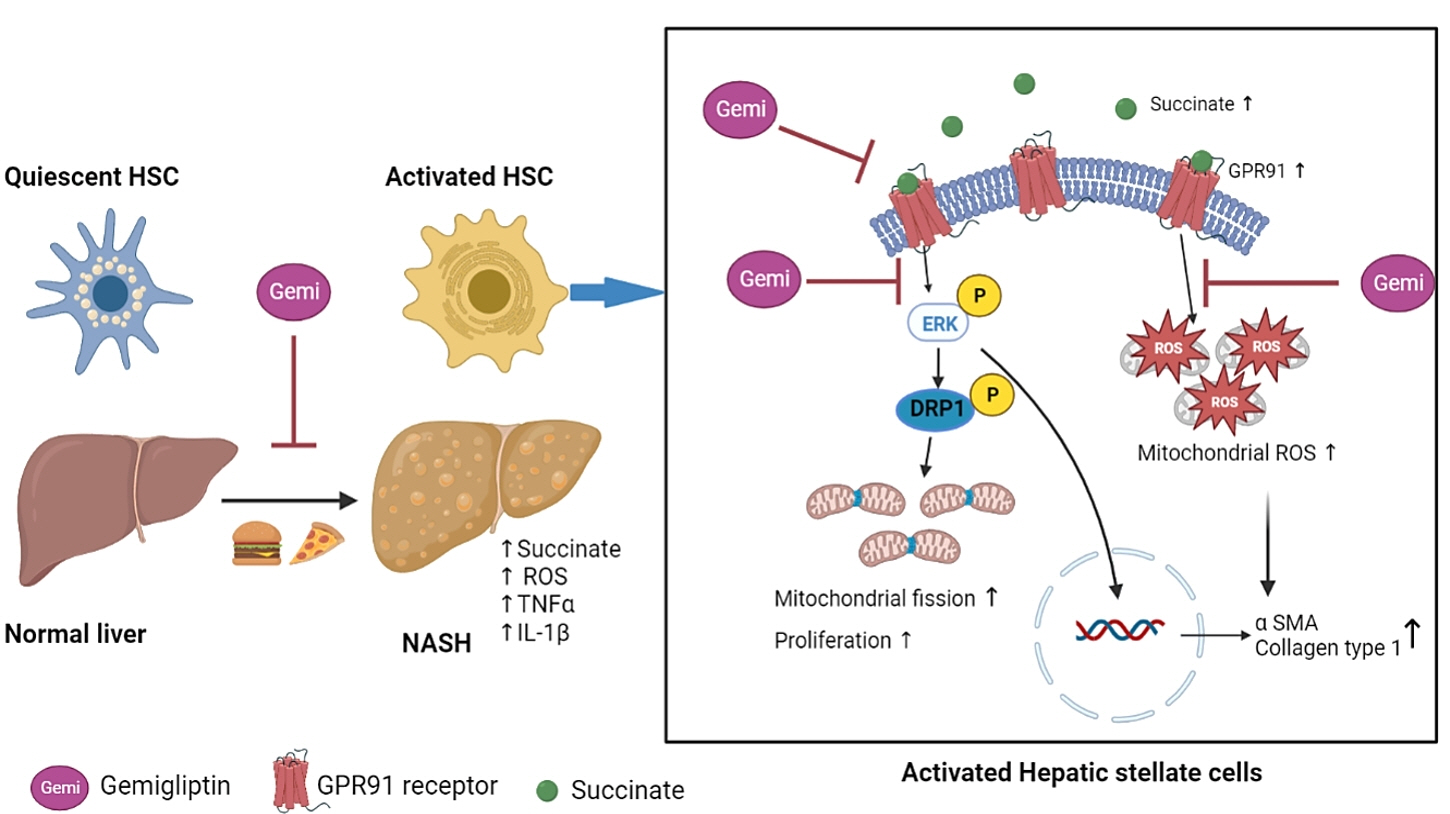
Gemigliptin Alleviates Succinate-Induced Hepatic Stellate Cell Activation by Ameliorating Mitochondrial Dysfunction
- Affiliations
-
- 1Department of Internal Medicine, Kangwon National University School of Medicine, Chuncheon, Korea
- KMID: 2537292
- DOI: http://doi.org/10.3803/EnM.2022.1530
Abstract
- Background
Dipeptidyl peptidase-4 inhibitors (DPP-4Is) are used clinically as oral antidiabetic agents. Although DPP-4Is are known to ameliorate liver fibrosis, the protective mechanism of DPP-4Is in liver fibrosis remains obscure. In this study, gemigliptin was used to investigate the potential of DPP-4Is to alleviate the progression of liver fibrosis.
Methods
To clarify the effects and mechanisms of gemigliptin, we conducted various experiments in LX-2 cells (immortalized human hepatic stellate cells [HSCs], the principal effectors of hepatic fibrogenesis), which were activated by succinate and exhibited elevated expression of α-smooth muscle actin, collagen type 1, and pro-inflammatory cytokines and increased cell proliferation. In vivo, we examined the effects and mechanisms of gemigliptin on a high-fat, high-cholesterol–induced mouse model of nonalcoholic steatohepatitis (NASH).
Results
Gemigliptin decreased the expression of fibrogenesis markers and reduced the abnormal proliferation of HSCs. In addition, gemigliptin reduced the succinate-induced production of mitochondrial reactive oxygen species (ROS), intracellular ROS, and mitochondrial fission in HSCs. Furthermore, in the mouse model of NASH-induced liver fibrosis, gemigliptin alleviated both liver fibrosis and mitochondrial dysfunction.
Conclusion
Gemigliptin protected against HSC activation and liver fibrosis by alleviating mitochondrial dysfunction and ROS production, indicating its potential as a strategy for preventing the development of liver disease.
Keyword
Figure
-
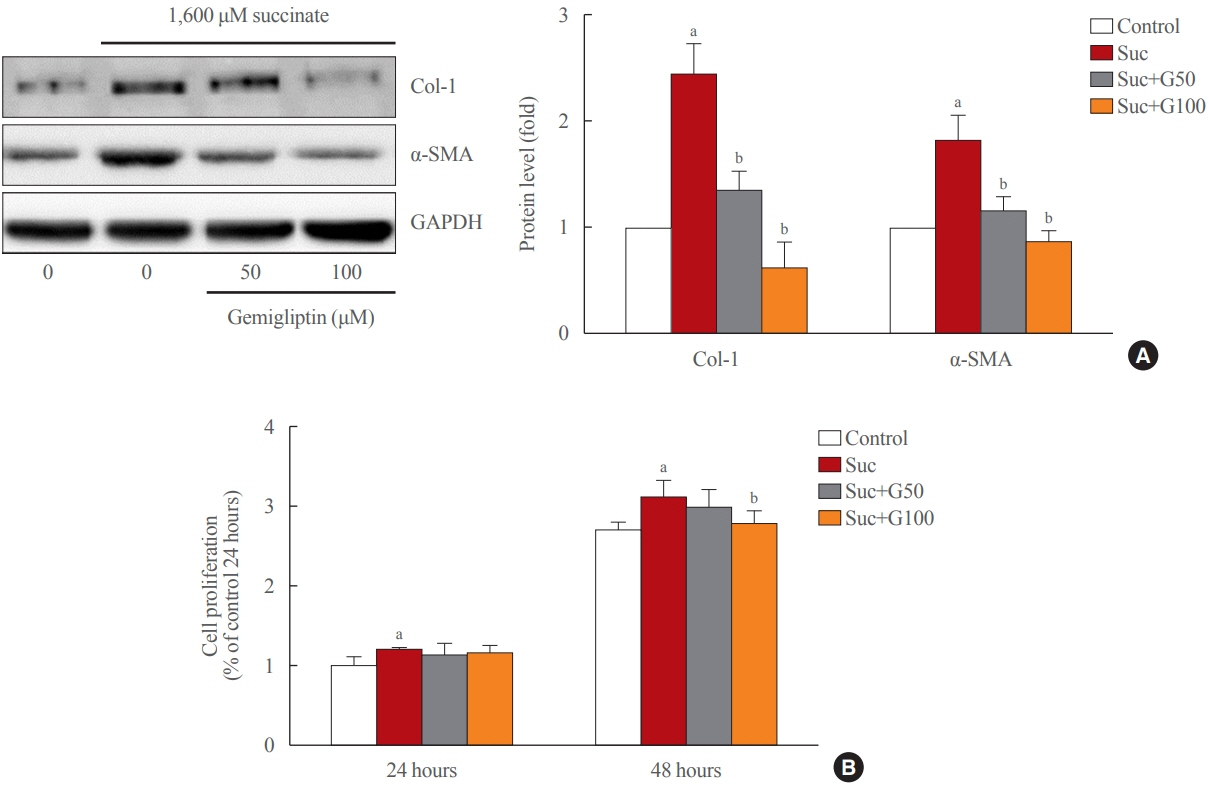
Fig. 1. Gemigliptin ameliorated succinate-induced hepatic stellate cell activation and proliferation. (A) LX-2 cells were treated with 1,600 μM succinate and co-treated with 50 or 100 μM gemigliptin for 24 hours. Western blot analysis of α-smooth muscle actin (α-SMA) and collagen type 1 (Col-1) expression in LX-2 cells. The band intensities in Western blot images were quantified using Image Lab and plotted to the right of the representative blot images. The protein levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. (B) The proliferation of LX-2 cells that were treated with 1,600 μM succinate and co-treated with 50 or 100 μM gemigliptin for 24 and 48 hours. aP<0.05 vs. control; bP<0.05 vs. succinate (mean±standard error of the mean, n=3).
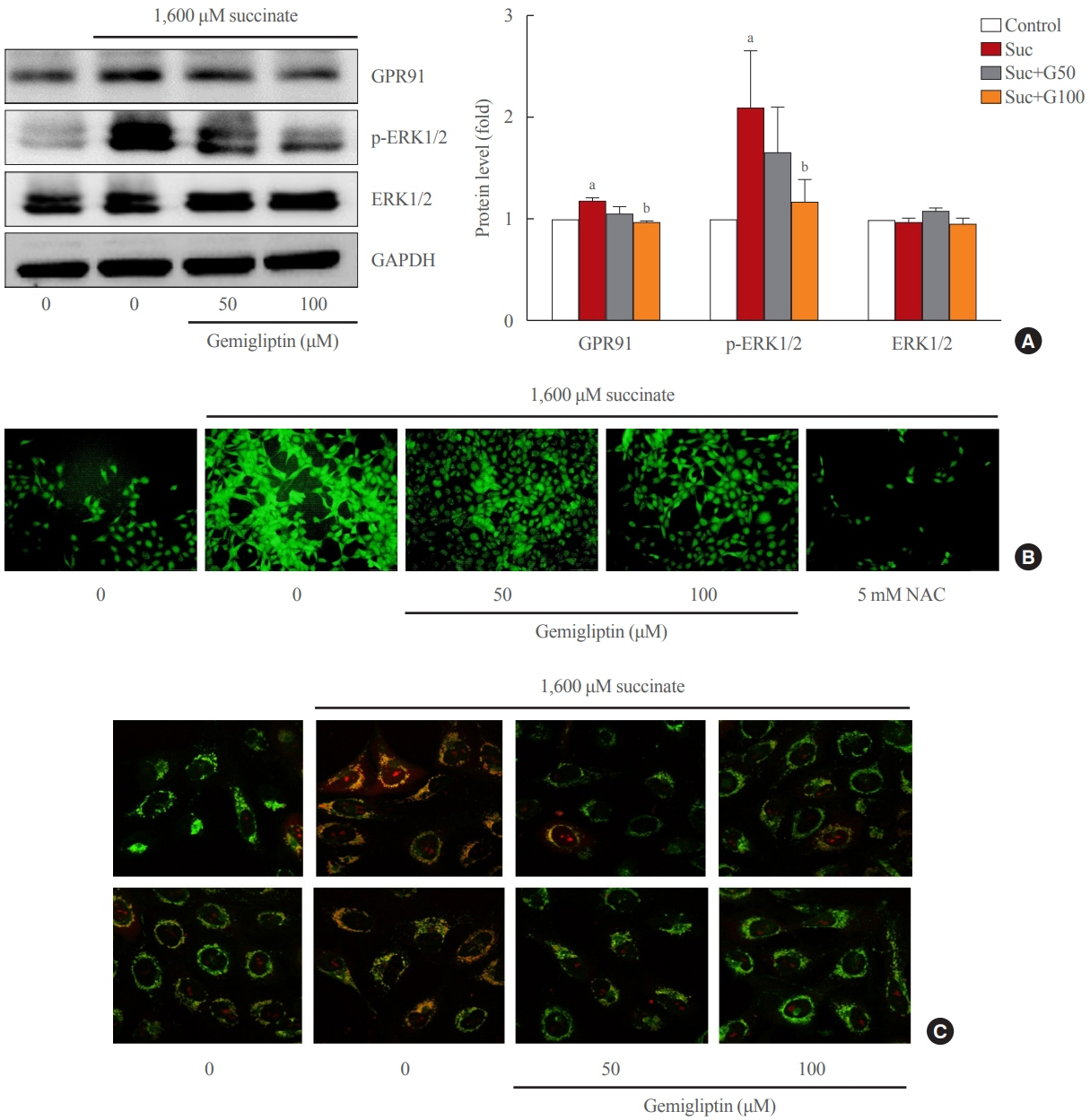
Fig. 2. Gemigliptin inhibited the succinate-G-protein coupled receptor 91 (GPR91) signaling pathway and reactive oxygen species (ROS) production in hepatic stellate cells. LX-2 cells were treated with 1,600 μM succinate and co-treated with 50 or 100 μM gemigliptin for 24 hours. (A) Western blot analysis of GPR91, phospho-extracellular signal-regulated kinase 1/2 (p-ERK1/2), and total ERK1/2 expression in LX-2 cells. The band intensities in Western blot images were quantified using Image Lab and plotted to the right of the representative blot images. The protein levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. (B) Determination of cellular ROS by the dichlorofluorescin diacetate (DCFDA) assay, fluorescence microscopic images of treated cells. (C) Mitochondrial localization of ROS in LX-2 cells in various treatment groups imaged using confocal scanning microscopy (green: mitochondria; red: MitoSOX). NAC, N-acetyl-L-cysteine. aP<0.05 vs. control; bP<0.05 vs. succinate (mean±standard error of the mean, n=3).
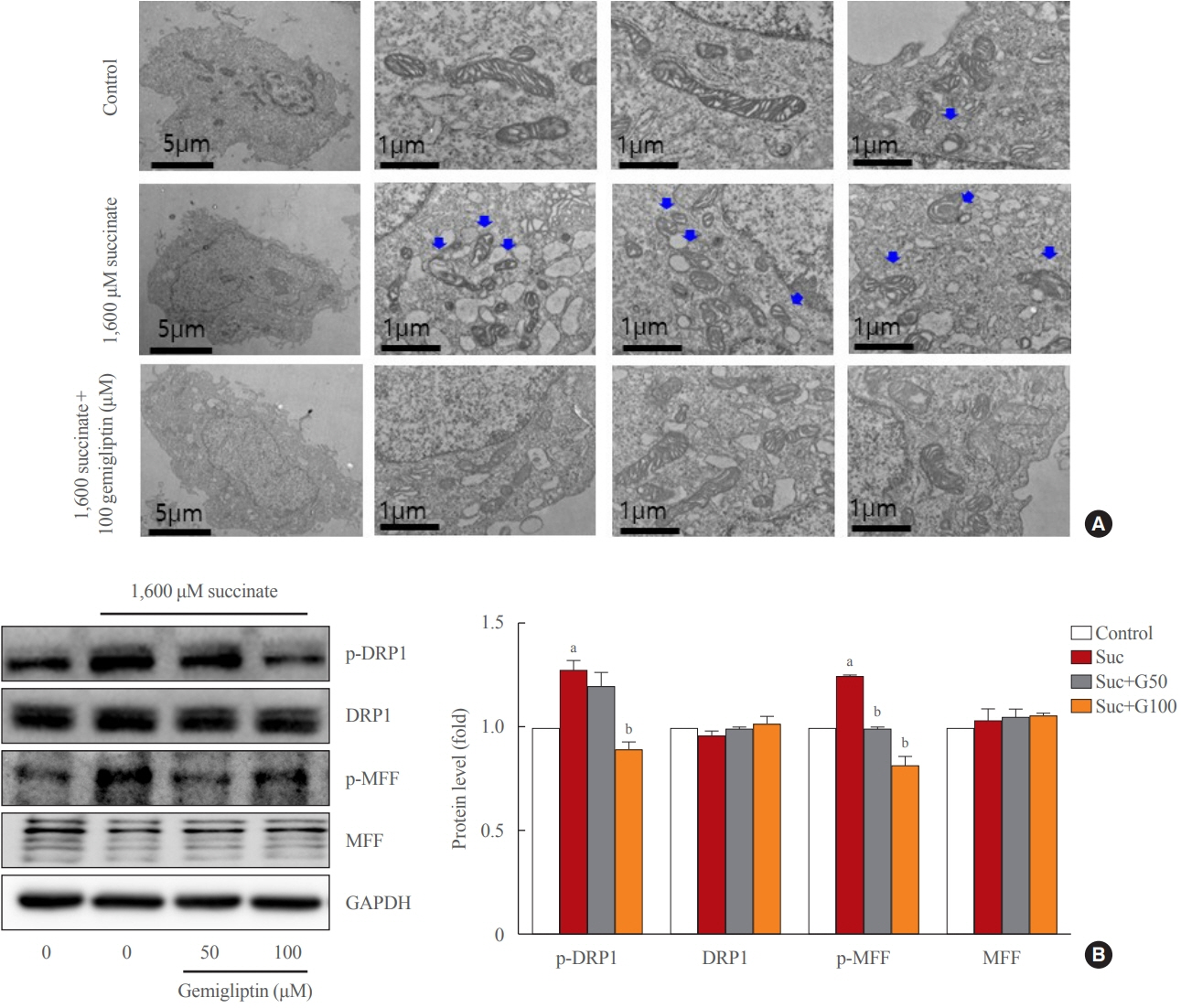
Fig. 3. Gemigliptin alleviated succinate-induced mitochondrial fragmentation. LX-2 cells were treated with 1,600 μM succinate and cotreated with 50 or 100 μM gemigliptin for 1 hour. (A) Mitochondrial fission was detected using transmission electron microscopy. The first left column shows a single LX-2 cell in the control and treatment groups (scale bar, 2 μm). The two panels in the right column are high-magnification images (scale bar, 1 μm). Blue arrows indicate mitochondria. (B) Western blot analysis of phospho-dynamin-related protein 1 (p-DRP1), phospho-mitochondrial fission factor (p-MFF), total DRP1, and total MFF expression in LX-2 cells. The band intensities in Western blot images were quantified using Image Lab and plotted to the right of the representative blot images. The protein levels were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) expression. aP<0.05 vs. control; bP<0.05 vs. succinate (mean±standard error of the mean, n=3).

Fig. 4. Gemigliptin attenuated liver fibrosis and inflammation in a high-fat, high-cholesterol diet mouse model. (A) H&E (left panels) and Masson’s trichrome staining (right panels) were performed to evaluate steatosis and liver fibrosis, at 4 weeks of feeding, respectively. The magnification of the image is shown. (B) H&E (left panels) and Masson’s trichrome staining (right panels) were performed to evaluate steatosis and liver fibrosis after 8 weeks of feeding, respectively. The magnification of the image is shown. (C) Western blot analysis of α-smooth muscle actin (α-SMA), and collagen type 1 (Col-1) expression in the liver from control, high-fat, high-cholesterol (HFHC) dietfed, and HFHC diet+gemigliptin-treated mice. Representative blots are shown above of the plots of the corresponding band intensities. (D) Interleukin-1β (IL-1β) and tumor necrosis factor-α (TNFα) mRNA expression levels in liver tissue were measured in the liver from control, HFHC diet-fed, and HFHC diet+gemigliptin-treated mice. aP<0.05 vs. control group; bP<0.05 vs. HFHC diet group (mean±standard error of the mean; control group [n=5], HFHC group [n=11], HFHC+gemigliptin group [n=11]).
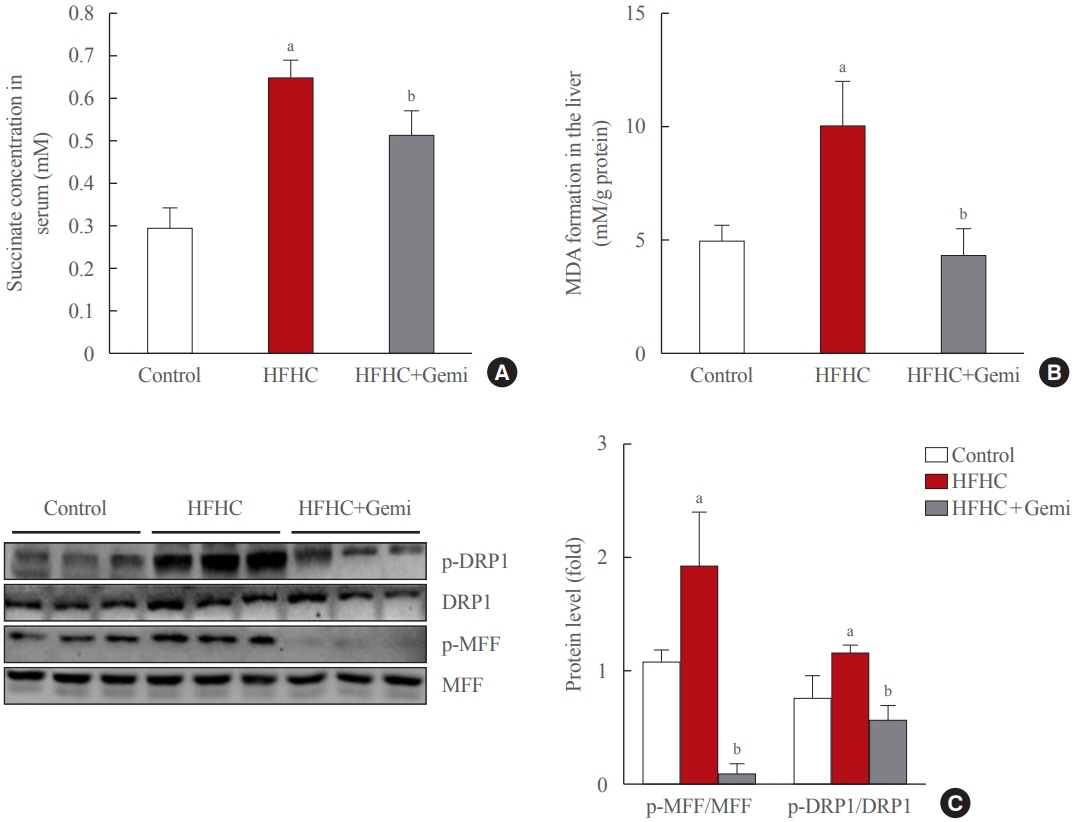
Fig. 5. Gemigliptin protected the liver from oxidative stress and mitochondrial fission. (A) Succinate concentration in mouse serum samples collected from the indicated groups. (B) malondialdehyde (MDA) formation was measured in the liver from control, high-fat high-cholesterol (HFHC) diet-fed, and HFHC diet+gemigliptin-treated mice. (C) Western blot analysis of phospho-dynamin-related protein 1 (p- DRP1), phospho-mitochondrial fission factor (p-MFF), total DRP1, and total MFF expression in the liver from control, HFHC diet-fed, and HFHC diet+gemigliptin-treated mice. Representative blots are shown to the left of the plots of the corresponding band intensities. aP<0.05 vs. control group; bP<0.05 vs. HFHC diet group (mean±standard error of the mean; control group [n=5], HFHC group [n=11], HFHC+ gemigliptin group [n=11]).
Cited by 2 articles
-
Insulin Resistance, Non-Alcoholic Fatty Liver Disease and Type 2 Diabetes Mellitus: Clinical and Experimental Perspective
Inha Jung, Dae-Jeong Koo, Won-Young Lee
Diabetes Metab J. 2024;48(3):327-339. doi: 10.4093/dmj.2023.0350.Irisin Attenuates Hepatic Stellate Cell Activation and Liver Fibrosis in Bile Duct Ligation Mice Model and Improves Mitochondrial Dysfunction
Thuy Linh Lai, So Young Park, Giang Nguyen, Phuc Thi Minh Pham, Seon Mee Kang, Jeana Hong, Jae-Ho Lee, Seung-Soon Im, Dae-Hee Choi, Eun-Hee Cho
Endocrinol Metab. 2024;39(6):908-920. doi: 10.3803/EnM.2024.1984.
Reference
-
1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005; 115:209–18.
Article2. Guo J, Friedman SL. Hepatic fibrogenesis. Semin Liver Dis. 2007; 27:413–26.
Article3. Ray I, Mahata SK, De RK. Obesity: an immunometabolic perspective. Front Endocrinol (Lausanne). 2016; 7:157.
Article4. Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013; 123:1902–10.
Article5. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008; 134:1655–69.
Article6. Cho EH. Succinate as a regulator of hepatic stellate cells in liver fibrosis. Front Endocrinol (Lausanne). 2018; 9:455.7. Li X, Xie L, Qu X, Zhao B, Fu W, Wu B, et al. GPR91, a critical signaling mechanism in modulating pathophysiologic processes in chronic illnesses. FASEB J. 2020; 34:13091–105.
Article8. Li YH, Woo SH, Choi DH, Cho EH. Succinate causes α-SMA production through GPR91 activation in hepatic stellate cells. Biochem Biophys Res Commun. 2015; 463:853–8.
Article9. Park SY, Le CT, Sung KY, Choi DH, Cho EH. Succinate induces hepatic fibrogenesis by promoting activation, proliferation, and migration, and inhibiting apoptosis of hepatic stellate cells. Biochem Biophys Res Commun. 2018; 496:673–8.
Article10. Cherry C, Thompson B, Saptarshi N, Wu J, Hoh J. 2016: a ‘Mitochondria’ odyssey. Trends Mol Med. 2016; 22:391–403.
Article11. Nasrallah CM, Horvath TL. Mitochondrial dynamics in the central regulation of metabolism. Nat Rev Endocrinol. 2014; 10:650–8.
Article12. Lu B. Mitochondrial dynamics and neurodegeneration. Dordrecht: Springer;2011. Chapter 2, Relationships between mitochondrial dynamics and bioenergetics. p. 47–68.13. Schrepfer E, Scorrano L. Mitofusins, from Mitochondria to metabolism. Mol Cell. 2016; 61:683–94.
Article14. Archer SL. Mitochondrial dynamics: mitochondrial fission and fusion in human diseases. N Engl J Med. 2013; 369:2236–51.
Article15. Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, et al. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010; 191:1141–58.
Article16. Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, et al. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015; 57:537–51.
Article17. Friedman SL. Liver fibrosis: from bench to bedside. J Hepatol. 2003; 38 Suppl 1:S38–53.18. Fallowfield JA. Therapeutic targets in liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011; 300:G709–15.
Article19. Jeong SW. Nonalcoholic fatty liver disease: a drug revolution is coming. Diabetes Metab J. 2020; 44:640–57.
Article20. Balaban YH, Korkusuz P, Simsek H, Gokcan H, Gedikoglu G, Pinar A, et al. Dipeptidyl peptidase IV (DDP IV) in NASH patients. Ann Hepatol. 2007; 6:242–50.
Article21. Miyazaki M, Kato M, Tanaka K, Tanaka M, Kohjima M, Nakamura K, et al. Increased hepatic expression of dipeptidyl peptidase-4 in non-alcoholic fatty liver disease and its association with insulin resistance and glucose metabolism. Mol Med Rep. 2012; 5:729–33.
Article22. Thornberry NA, Gallwitz B. Mechanism of action of inhibitors of dipeptidyl-peptidase-4 (DPP-4). Best Pract Res Clin Endocrinol Metab. 2009; 23:479–86.
Article23. Gorrell MD. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin Sci (Lond). 2005; 108:277–92.
Article24. Itou M, Kawaguchi T, Taniguchi E, Sata M. Dipeptidyl peptidase-4: a key player in chronic liver disease. World J Gastroenterol. 2013; 19:2298–306.
Article25. Kaji K, Yoshiji H, Ikenaka Y, Noguchi R, Aihara Y, Douhara A, et al. Dipeptidyl peptidase-4 inhibitor attenuates hepatic fibrosis via suppression of activated hepatic stellate cell in rats. J Gastroenterol. 2014; 49:481–91.
Article26. Kawakubo M, Tanaka M, Ochi K, Watanabe A, Saka-Tanaka M, Kanamori Y, et al. Dipeptidyl peptidase-4 inhibition prevents nonalcoholic steatohepatitis-associated liver fibrosis and tumor development in mice independently of its antidiabetic effects. Sci Rep. 2020; 10:983.
Article27. Pintana H, Apaijai N, Chattipakorn N, Chattipakorn SC. DPP-4 inhibitors improve cognition and brain mitochondrial function of insulin-resistant rats. J Endocrinol. 2013; 218:1–11.
Article28. Gandhi CR. Oxidative stress and hepatic stellate cells: a paradoxical relationship. Trends Cell Mol Biol. 2012; 7:1–10.29. Astiarraga B, Martinez L, Ceperuelo-Mallafre V, Llaurado G, Terron-Puig M, Rodriguez MM, et al. Impaired succinate response to a mixed meal in obesity and type 2 diabetes is normalized after metabolic surgery. Diabetes Care. 2020; 43:2581–7.
Article30. Fernandez-Veledo S, Vendrell J. Gut microbiota-derived succinate: friend or foe in human metabolic diseases? Rev Endocr Metab Disord. 2019; 20:439–47.
Article31. Ceperuelo-Mallafre V, Llaurado G, Keiran N, Benaiges E, Astiarraga B, Martinez L, et al. Preoperative circulating succinate levels as a biomarker for diabetes remission after bariatric surgery. Diabetes Care. 2019; 42:1956–65.
Article32. van Diepen JA, Robben JH, Hooiveld GJ, Carmone C, Alsady M, Boutens L, et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia. 2017; 60:1304–13.
Article33. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013; 496:238–42.
Article34. Tretter L, Patocs A, Chinopoulos C. Succinate, an intermediate in metabolism, signal transduction, ROS, hypoxia, and tumorigenesis. Biochim Biophys Acta. 2016; 1857:1086–101.
Article35. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan Gprotein-coupled receptors. Nature. 2004; 429:188–93.
Article36. Guo Y, Cho SW, Saxena D, Li X. Multifaceted actions of succinate as a signaling transmitter vary with its cellular locations. Endocrinol Metab (Seoul). 2020; 35:36–43.
Article37. Vakifahmetoglu-Norberg H, Ouchida AT, Norberg E. The role of mitochondria in metabolism and cell death. Biochem Biophys Res Commun. 2017; 482:426–31.
Article38. Ezhilarasan D. Oxidative stress is bane in chronic liver diseases: clinical and experimental perspective. Arab J Gastroenterol. 2018; 19:56–64.
Article39. Kleele T, Rey T, Winter J, Zaganelli S, Mahecic D, Perreten Lambert H, et al. Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature. 2021; 593:435–9.
Article40. Lu YT, Li LZ, Yang YL, Yin X, Liu Q, Zhang L, et al. Succinate induces aberrant mitochondrial fission in cardiomyocytes through GPR91 signaling. Cell Death Dis. 2018; 9:672.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Phloretin Ameliorates Succinate-Induced Liver Fibrosis by Regulating Hepatic Stellate Cells
- Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis
- Irisin Attenuates Hepatic Stellate Cell Activation and Liver Fibrosis in Bile Duct Ligation Mice Model and Improves Mitochondrial Dysfunction
- Control of Mitochondrial Dynamics by Fas-induced Caspase-8 Activation in Hippocampal Neurons
- Experimental Animal Models of Hepatic Fibrosis
