Neonatal Med.
2021 Feb;28(1):59-63. 10.5385/nm.2021.28.1.59.
Congenital Long QT Syndrome Type 8 Characterized by Fetal Onset of Bradycardia and 2:1 Atrioventricular Block
- Affiliations
-
- 1Department of Pediatrics, Pusan National University Children’s Hospital, Yangsan, Korea
- KMID: 2513361
- DOI: http://doi.org/10.5385/nm.2021.28.1.59
Abstract
- An important, albeit rare, cause of fetal bradycardia is long QT syndrome (LQTS). Congenital LQTS is an ion channelopathy caused by mutations in genes encoding cardiac ion channel proteins. Fetal onset of LQTS imposes high risk of life-threatening tachyarrhythmias and sudden cardiac death. Here, we report the case of a female newborn with fetal onset of bradycardia and a 2:1 atrioventricular (AV) block. After birth, a 12-lead electrocardiogram (ECG) revealed bradycardia with QT prolongation of a corrected QT (QTc) interval of 680 ms and pseudo 2:1 AV block. Genetic testing identified a heterozygous Gly402Ser (c.1204G>A) mutation in CACNA1C, confirming the diagnosis of LQTS type 8 (LQT8). The patient received propranolol at a daily dose of 2 mg/kg. Mexiletine was subsequently administered owing to the sustained prolongation of the QT interval and pseudo 2:1 AV block. One week after mexiletine inception, the ECG still showed QT interval prolongation (QTc, 632 ms), but no AV block was observed. There were no life-threatening tachyarrhythmias in a follow-up period of 13 months.
Figure
-
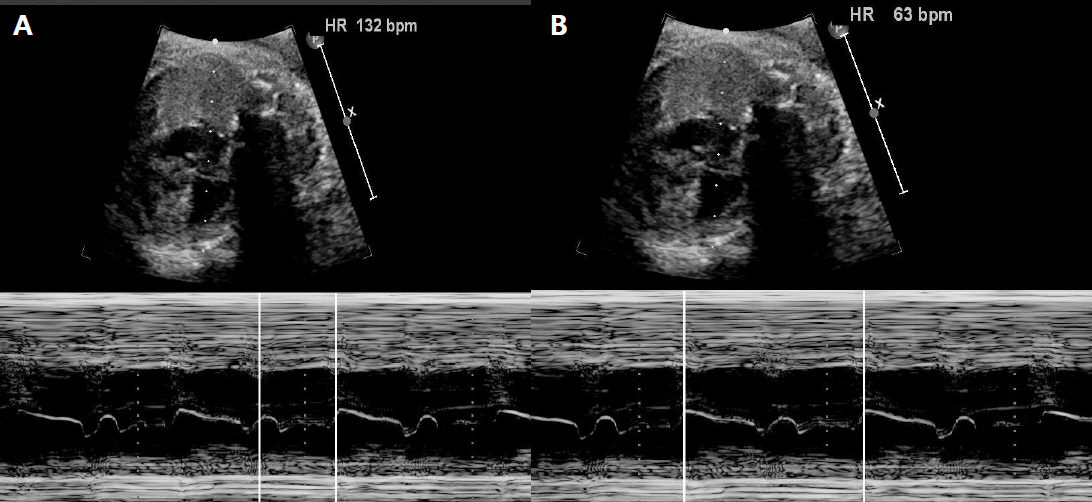
Figure 1. Fetal M-mode echocardiogram showed a 2:1 atrioventricular block. The atrial rate (A) was 132 bpm and the ventricular rate (B) was 63 bpm.

Figure 2. Postnatal 12-lead electrocardiogram showed bradycardia. The heart rate was 68 beats/min with QT prolongation of a corrected QT interval (QTc) of 680 ms (using Bazett formula) and a pseudo 2:1 atrioventricular block.

Figure 3. Electrocardiogram continued to show QT interval prolongation (QTc 632 ms) but revealed no atrioventricular block after persistent mexiletine administration.
Reference
-
1. Maeno Y, Hirose A, Kanbe T, Hori D. Fetal arrhythmia: prenatal diagnosis and perinatal management. J Obstet Gynaecol Res. 2009; 35:623–9.2. Anuwutnavin S, Wanitpongpan P, Chungsomprasong P, Soongswang J, Srisantiroj N, Wataganara T. Fetal long QT syndrome manifested as atrioventricular block and ventricular tachycardia: a case report and a review of the literature. Pediatr Cardiol. 2013; 34:1955–62.3. Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013; 62:169–80.4. Skinner JR, Winbo A, Abrams D, Vohra J, Wilde AA. Channelopathies that lead to sudden cardiac death: clinical and genetic aspects. Heart Lung Circ. 2019; 28:22–30.5. Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004; 119:19–31.6. Fukuyama M, Wang Q, Kato K, Ohno S, Ding WG, Toyoda F, et al. Long QT syndrome type 8: novel CACNA1C mutations causing QT prolongation and variant phenotypes. Europace. 2014; 16:1828–37.7. Wemhoner K, Friedrich C, Stallmeyer B, Coffey AJ, Grace A, Zumhagen S, et al. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J Mol Cell Cardiol. 2015; 80:186–95.8. Fukuyama M, Ohno S, Ozawa J, Kato K, Makiyama T, Nakagawa Y, et al. High prevalence of late-appearing T-wave in patients with long QT syndrome type 8. Circ J. 2020; 84:559–68.9. Adler A, Novelli V, Amin AS, Abiusi E, Care M, Nannenberg EA, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation. 2020; 141:418–28.10. Cuneo BF, Etheridge SP, Horigome H, Sallee D, Moon-Grady A, Weng HY, et al. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: risk stratification of perinatal long-QT syndrome. Circ Arrhythm Electrophysiol. 2013; 6:946–51.11. Dufendach KA, Timothy K, Ackerman MJ, Blevins B, Pflaumer A, Etheridge S, et al. Clinical outcomes and modes of death in timothy syndrome: a multicenter international study of a rare disorder. JACC Clin Electrophysiol. 2018; 4:459–66.12. Rosenbaum MB, Acunzo RS. Pseudo 2:1 atrioventricular block and T wave alternans in the long QT syndromes. J Am Coll Cardiol. 1991; 18:1363–6.13. Mazzanti A, Maragna R, Faragli A, Monteforte N, Bloise R, Memmi M, et al. Gene-specific therapy with mexiletine reduces arrhythmic events in patients with long QT syndrome type 3. J Am Coll Cardiol. 2016; 67:1053–8.14. Okuwaki H, Kato Y, Lin L, Nozaki Y, Takahashi-Igari M, Horigome H. Mexiletine infusion challenge test for neonatal long QT syndrome with 2:1 atrioventricular block. J Arrhythm. 2019; 35:685–8.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of Congenital Long OT Syndrome with Pseudo - Atrioventricular Block
- A Case of Idiopathic Long QT Syndrome with 2:1 Atrioventricular Block
- Two Cases of 2:1 Atrioventricular Block in Infants with Idiopathic Long QT Syndrome
- One Case of Congenital Complete Atrioventricular Block Diagnosed by Fetal Echocardiography
- A Case of Neonatal Lupus Syndrome with Congenital Heart Block
