Endocrinol Metab.
2020 Sep;35(3):494-506. 10.3803/EnM.2020.302.
WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System
- Affiliations
-
- 1Cell Biology Research Centre, Molecular and Clinical Sciences Research Institute, St. George’s, University of London, London, UK
- KMID: 2507998
- DOI: http://doi.org/10.3803/EnM.2020.302
Abstract
- WD40-repeat (WDR)-containing proteins constitute an evolutionarily conserved large protein family with a broad range of biological functions. In human proteome, WDR makes up one of the most abundant protein-protein interaction domains. Members of the WDR protein family play important roles in nearly all major cellular signalling pathways. Mutations of WDR proteins have been associated with various human pathologies including neurological disorders, cancer, obesity, ciliopathies and endocrine disorders. This review provides an updated overview of the biological functions of WDR proteins and their mutations found in congenital disorders. We also highlight the significant role of WDR proteins in ciliopathies and endocrine disorders. The new insights may help develop therapeutic approaches targeting WDR motifs.
Keyword
Figure
-
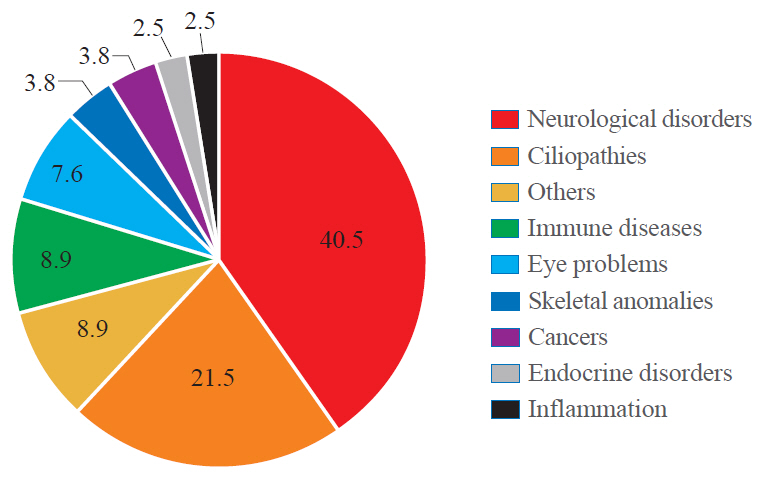
Fig. 1. A chart indicating the relevant prevalence of human diseases associated with WD40-repeat (WDR) proteins. The data are based on the entries in Online Mendelian Inheritance in Man (OMIM). Total 79 out of 360 WDR proteins have been linked with disease categories as indicated. The ‘others’ category includes multi-organ defects, liver failure, cardiovascular defects and embryonic lethality. The full list of WDR proteins assessed are included in Supplementary Table S1.
Reference
-
1. Li D, Roberts R. WD-repeat proteins: structure characteristics, biological function, and their involvement in human diseases. Cell Mol Life Sci. 2001; 58:2085–97.2. Stirnimann CU, Petsalaki E, Russell RB, Muller CW. WD40 proteins propel cellular networks. Trends Biochem Sci. 2010; 35:565–74.
Article3. Wall MA, Coleman DE, Lee E, Iniguez-Lluhi JA, Posner BA, Gilman AG, et al. The structure of the G protein heterotrimer Gi alpha 1 beta 1 gamma 2. Cell. 1995; 83:1047–58.4. Jain BP, Pandey S. WD40 repeat proteins: signalling scaffold with diverse functions. Protein J. 2018; 37:391–406.
Article5. Yu L, Gaitatzes C, Neer E, Smith TF. Thirty-plus functional families from a single motif. Protein Sci. 2000; 9:2470–6.
Article6. Schapira M, Tyers M, Torrent M, Arrowsmith CH. WD40 repeat domain proteins: a novel target class? Nat Rev Drug Discov. 2017; 16:773–86.
Article7. Pashkova N, Gakhar L, Winistorfer SC, Yu L, Ramaswamy S, Piper RC. WD40 repeat propellers define a ubiquitin-binding domain that regulates turnover of F box proteins. Mol Cell. 2010; 40:433–43.
Article8. Linossi EM, Nicholson SE. The SOCS box-adapting proteins for ubiquitination and proteasomal degradation. IUBMB Life. 2012; 64:316–23.
Article9. Wu G, Xu G, Schulman BA, Jeffrey PD, Harper JW, Pavletich NP. Structure of a beta-TrCP1-Skp1-beta-catenin complex: destruction motif binding and lysine specificity of the SCF(beta-TrCP1) ubiquitin ligase. Mol Cell. 2003; 11:1445–56.10. Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, et al. WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci U S A. 2017; 114:E9308–17.
Article11. Lystad AH, Simonsen A. Phosphoinositide-binding proteins in autophagy. FEBS Lett. 2016; 590:2454–68.
Article12. von Kleist L, Stahlschmidt W, Bulut H, Gromova K, Puchkov D, Robertson MJ, et al. Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell. 2011; 146:471–84.
Article13. Wang X, Huang Y, Yan M, Li J, Ding C, Jin H, et al. Molecular spectrum of excision repair cross-complementation group 8 gene defects in Chinese patients with Cockayne syndrome type A. Sci Rep. 2017; 7:13686.
Article14. Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, et al. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006; 20:1429–34.
Article15. van der Weegen Y, Golan-Berman H, Mevissen TET, Apelt K, Gonzalez-Prieto R, Goedhart J, et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat Commun. 2020; 11:2104.
Article16. Davis RJ, Welcker M, Clurman BE. Tumor suppression by the Fbw7 ubiquitin ligase: mechanisms and opportunities. Cancer Cell. 2014; 26:455–64.
Article17. Siu KT, Rosner MR, Minella AC. An integrated view of cyclin E function and regulation. Cell Cycle. 2012; 11:57–64.
Article18. Cronshaw JM, Matunis MJ. The nuclear pore complex protein ALADIN is mislocalized in triple A syndrome. Proc Natl Acad Sci U S A. 2003; 100:5823–7.
Article19. Kumar A, Sharma P, Gomar-Alba M, Shcheprova Z, Daulny A, Sanmartin T, et al. Daughter-cell-specific modulation of nuclear pore complexes controls cell cycle entry during asymmetric division. Nat Cell Biol. 2018; 20:432–42.
Article20. Alexandrov A, Martzen MR, Phizicky EM. Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA. 2002; 8:1253–66.
Article21. Lin S, Liu Q, Lelyveld VS, Choe J, Szostak JW, Gregory RI. Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-renewal and differentiation. Mol Cell. 2018; 71:244–55.
Article22. Cole DG. The intraflagellar transport machinery of Chlamydomonas reinhardtii. Traffic. 2003; 4:435–42.23. Hao L, Scholey JM. Intraflagellar transport at a glance. J Cell Sci. 2009; 122(Pt 7):889–92.
Article24. Schoenmakers N, Alatzoglou KS, Chatterjee VK, Dattani MT. Recent advances in central congenital hypothyroidism. J Endocrinol. 2015; 227:R51–71.
Article25. van Nocker S, Ludwig P. The WD-repeat protein superfamily in Arabidopsis: conservation and divergence in structure and function. BMC Genomics. 2003; 4:50.
Article26. Feng Y, Zhang C, Luo Q, Wei X, Jiang B, Zhu H, et al. A novel WD-repeat protein, WDR26, inhibits apoptosis of cardiomyocytes induced by oxidative stress. Free Radic Res. 2012; 46:777–84.
Article27. Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, et al. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004; 117:527–39.28. Van Roosbroeck K, Cox L, Tousseyn T, Lahortiga I, Gielen O, Cauwelier B, et al. JAK2 rearrangements, including the novel SEC31A-JAK2 fusion, are recurrent in classical Hodgkin lymphoma. Blood. 2011; 117:4056–64.
Article29. Proikas-Cezanne T, Takacs Z, Donnes P, Kohlbacher O. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015; 128:207–17.30. Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007; 8:880–93.
Article31. Malicki JJ, Johnson CA. The cilium: cellular antenna and central processing unit. Trends Cell Biol. 2017; 27:126–40.
Article32. Wemmer KA, Marshall WF. Flagellar motility: all pull together. Curr Biol. 2004; 14:R992–3.
Article33. Smith EF, Yang P. The radial spokes and central apparatus: mechano-chemical transducers that regulate flagellar motility. Cell Motil Cytoskeleton. 2004; 57:8–17.
Article34. Sung CH, Leroux MR. The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat Cell Biol. 2013; 15:1387–97.
Article35. Taschner M, Lorentzen E. The intraflagellar transport machinery. Cold Spring Harb Perspect Biol. 2016; 8:a028092.
Article36. Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002; 3:813–25.
Article37. Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol. 2003; 19:423–43.
Article38. Singla V, Reiter JF. The primary cilium as the cell’s antenna: signaling at a sensory organelle. Science. 2006; 313:629–33.
Article39. Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, Aguilar A, Schneider-Maunoury S, et al. Hedgehog signaling and primary cilia are required for the formation of adult neural stem cells. Nat Neurosci. 2008; 11:277–84.
Article40. Christensen ST, Clement CA, Satir P, Pedersen LB. Primary cilia and coordination of receptor tyrosine kinase (RTK) signalling. J Pathol. 2012; 226:172–84.
Article41. Schou KB, Pedersen LB, Christensen ST. Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 2015; 16:1099–113.42. Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010; 11:331–44.
Article43. Wheway G, Nazlamova L, Hancock JT. Signaling through the primary cilium. Front Cell Dev Biol. 2018; 6:8.
Article44. Baker K, Beales PL. Making sense of cilia in disease: the human ciliopathies. Am J Med Genet C Semin Med Genet. 2009; 151C:281–95.
Article45. Waters AM, Beales PL. Ciliopathies: an expanding disease spectrum. Pediatr Nephrol. 2011; 26:1039–56.
Article46. Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011; 364:1533–43.
Article47. Dobyns WB, Curry CJ, Hoyme HE, Turlington L, Ledbetter DH. Clinical and molecular diagnosis of Miller-Dieker syndrome. Am J Hum Genet. 1991; 48:584–94.48. Cardoso C, Leventer RJ, Ward HL, Toyo-Oka K, Chung J, Gross A, et al. Refinement of a 400-kb critical region allows genotypic differentiation between isolated lissencephaly, Miller-Dieker syndrome, and other phenotypes secondary to deletions of 17p13.3. Am J Hum Genet. 2003; 72:918–30.
Article49. Lo Nigro C, Chong CS, Smith AC, Dobyns WB, Carrozzo R, Ledbetter DH. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997; 6:157–64.
Article50. Cardoso C, Leventer RJ, Matsumoto N, Kuc JA, Ramocki MB, Mewborn SK, et al. The location and type of mutation predict malformation severity in isolated lissencephaly caused by abnormalities within the LIS1 gene. Hum Mol Genet. 2000; 9:3019–28.
Article51. Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993; 270:2838–42.
Article52. Sapir T, Cahana A, Seger R, Nekhai S, Reiner O. LIS1 is a microtubule-associated phosphoprotein. Eur J Biochem. 1999; 265:181–8.
Article53. Jimenez-Mateos EM, Wandosell F, Reiner O, Avila J, Gonzalez-Billault C. Binding of microtubule-associated protein 1B to LIS1 affects the interaction between dynein and LIS1. Biochem J. 2005; 389(Pt 2):333–41.
Article54. Yigit G, Wieczorek D, Bogershausen N, Beleggia F, Moller-Hartmann C, Altmuller J, et al. A syndrome of microcephaly, short stature, polysyndactyly, and dental anomalies caused by a homozygous KATNB1 mutation. Am J Med Genet A. 2016; 170:728–33.55. Mishra-Gorur K, Caglayan AO, Schaffer AE, Chabu C, Henegariu O, Vonhoff F, et al. Mutations in KATNB1 cause complex cerebral malformations by disrupting asymmetrically dividing neural progenitors. Neuron. 2014; 84:1226–39.
Article56. McNally KP, Bazirgan OA, McNally FJ. Two domains of p80 katanin regulate microtubule severing and spindle pole targeting by p60 katanin. J Cell Sci. 2000; 113(Pt 9):1623–33.
Article57. McNally FJ, Okawa K, Iwamatsu A, Vale RD. Katanin, the microtubule-severing ATPase, is concentrated at centrosomes. J Cell Sci. 1996; 109(Pt 3):561–7.
Article58. Hartman JJ, Mahr J, McNally K, Okawa K, Iwamatsu A, Thomas S, et al. Katanin, a microtubule-severing protein, is a novel AAA ATPase that targets to the centrosome using a WD40-containing subunit. Cell. 1998; 93:277–87.
Article59. Ahmad FJ, Yu W, McNally FJ, Baas PW. An essential role for katanin in severing microtubules in the neuron. J Cell Biol. 1999; 145:305–15.
Article60. Gandhi PN, Wang X, Zhu X, Chen SG, Wilson-Delfosse AL. The Roc domain of leucine-rich repeat kinase 2 is sufficient for interaction with microtubules. J Neurosci Res. 2008; 86:1711–20.
Article61. Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, et al. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012; 491:603–7.
Article62. Kluss JH, Mamais A, Cookson MR. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem Soc Trans. 2019; 47:651–61.
Article63. Cirnaru MD, Marte A, Belluzzi E, Russo I, Gabrielli M, Longo F, et al. LRRK2 kinase activity regulates synaptic vesicle trafficking and neurotransmitter release through modulation of LRRK2 macro-molecular complex. Front Mol Neurosci. 2014; 7:49.
Article64. Farrer MJ, Stone JT, Lin CH, Dachsel JC, Hulihan MM, Haugarvoll K, et al. Lrrk2 G2385R is an ancestral risk factor for Parkinson’s disease in Asia. Parkinsonism Relat Disord. 2007; 13:89–92.
Article65. Funayama M, Li Y, Tomiyama H, Yoshino H, Imamichi Y, Yamamoto M, et al. Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport. 2007; 18:273–5.
Article66. Tan EK, Zhao Y, Skipper L, Tan MG, Di Fonzo A, Sun L, et al. The LRRK2 Gly2385Arg variant is associated with Parkinson’s disease: genetic and functional evidence. Hum Genet. 2007; 120:857–63.
Article67. Carrion MDP, Marsicano S, Daniele F, Marte A, Pischedda F, Di Cairano E, et al. The LRRK2 G2385R variant is a partial loss-of-function mutation that affects synaptic vesicle trafficking through altered protein interactions. Sci Rep. 2017; 7:5377.
Article68. Scheibye-Knudsen M, Tseng A, Borch Jensen M, ScheibyeAlsing K, Fang EF, Iyama T, et al. Cockayne syndrome group A and B proteins converge on transcription-linked resolution of non-B DNA. Proc Natl Acad Sci U S A. 2016; 113:12502–7.
Article69. Allgrove J, Clayden GS, Grant DB, Macaulay JC. Familial glucocorticoid deficiency with achalasia of the cardia and deficient tear production. Lancet. 1978; 1:1284–6.
Article70. Grant DB, Barnes ND, Dumic M, Ginalska-Malinowska M, Milla PJ, von Petrykowski W, et al. Neurological and adrenal dysfunction in the adrenal insufficiency/alacrima/achalasia (3A) syndrome. Arch Dis Child. 1993; 68:779–82.
Article71. Handschug K, Sperling S, Yoon SJ, Hennig S, Clark AJ, Huebner A. Triple A syndrome is caused by mutations in AAAS, a new WD-repeat protein gene. Hum Mol Genet. 2001; 10:283–90.
Article72. Huebner A, Kaindl AM, Knobeloch KP, Petzold H, Mann P, Koehler K. The triple A syndrome is due to mutations in ALADIN, a novel member of the nuclear pore complex. Endocr Res. 2004; 30:891–9.
Article73. Hirano M, Furiya Y, Asai H, Yasui A, Ueno S. ALADINI482S causes selective failure of nuclear protein import and hypersensitivity to oxidative stress in triple A syndrome. Proc Natl Acad Sci U S A. 2006; 103:2298–303.74. Storr HL, Kind B, Parfitt DA, Chapple JP, Lorenz M, Koehler K, et al. Deficiency of ferritin heavy-chain nuclear import in triple a syndrome implies nuclear oxidative damage as the primary disease mechanism. Mol Endocrinol. 2009; 23:2086–94.
Article75. Shaheen R, Abdel-Salam GM, Guy MP, Alomar R, Abdel-Hamid MS, Afifi HH, et al. Mutation in WDR4 impairs tRNA m(7)G46 methylation and causes a distinct form of microcephalic primordial dwarfism. Genome Biol. 2015; 16:210.
Article76. Trimouille A, Lasseaux E, Barat P, Deiller C, Drunat S, Rooryck C, et al. Further delineation of the phenotype caused by biallelic variants in the WDR4 gene. Clin Genet. 2018; 93:374–7.
Article77. Braun DA, Shril S, Sinha A, Schneider R, Tan W, Ashraf S, et al. Mutations in WDR4 as a new cause of Galloway-Mowat syndrome. Am J Med Genet A. 2018; 176:2460–5.78. Pezzella M, Yeghiazaryan NS, Veggiotti P, Bettinelli A, Giudizioso G, Zara F, et al. Galloway-Mowat syndrome: an early-onset progressive encephalopathy with intractable epilepsy associated to renal impairment. Two novel cases and review of literature. Seizure. 2010; 19:132–5.
Article79. Krishnamurthy S, Rajesh NG, Ramesh A, Zenker M. Infantile nephrotic syndrome with microcephaly and global developmental delay: the Galloway Mowat Syndrome. Indian J Pediatr. 2012; 79:1087–90.
Article80. Jinks RN, Puffenberger EG, Baple E, Harding B, Crino P, Fogo AB, et al. Recessive nephrocerebellar syndrome on the Galloway-Mowat syndrome spectrum is caused by homozygous protein-truncating mutations of WDR73. Brain. 2015; 138(Pt 8):2173–90.
Article81. Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet. 2014; 95:637–48.
Article82. Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007; 39:162–4.
Article83. Cortellino S, Wang C, Wang B, Bassi MR, Caretti E, Champeval D, et al. Defective ciliogenesis, embryonic lethality and severe impairment of the Sonic Hedgehog pathway caused by inactivation of the mouse complex A intraflagellar transport gene Ift122/Wdr10, partially overlapping with the DNA repair gene Med1/Mbd4. Dev Biol. 2009; 325:225–37.
Article84. Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011; 89:634–43.
Article85. Mill P, Lockhart PJ, Fitzpatrick E, Mountford HS, Hall EA, Reijns MA, et al. Human and mouse mutations in WDR35 cause short-rib polydactyly syndromes due to abnormal ciliogenesis. Am J Hum Genet. 2011; 88:508–15.
Article86. Arts HH, Bongers EM, Mans DA, van Beersum SE, Oud MM, Bolat E, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011; 48:390–5.
Article87. Schmidts M, Frank V, Eisenberger T, Al Turki S, Bizet AA, Antony D, et al. Combined NGS approaches identify mutations in the intraflagellar transport gene IFT140 in skeletal ciliopathies with early progressive kidney disease. Hum Mutat. 2013; 34:714–24.88. Davis EE, Zhang Q, Liu Q, Diplas BH, Davey LM, Hartley J, et al. TTC21B contributes both causal and modifying alleles across the ciliopathy spectrum. Nat Genet. 2011; 43:189–96.
Article89. Cavalcanti DP, Huber C, Sang KH, Baujat G, Collins F, Delezoide AL, et al. Mutation in IFT80 in a fetus with the phenotype of Verma-Naumoff provides molecular evidence for Jeune-Verma-Naumoff dysplasia spectrum. J Med Genet. 2011; 48:88–92.
Article90. Halbritter J, Bizet AA, Schmidts M, Porath JD, Braun DA, Gee HY, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013; 93:915–25.
Article91. Zhang W, Taylor SP, Nevarez L, Lachman RS, Nickerson DA, Bamshad M, et al. IFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome. Hum Mol Genet. 2016; 25:4012–20.92. Duran I, Taylor SP, Zhang W, Martin J, Forlenza KN, Spiro RP, et al. Destabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a spectrum of short-rib polydactyly syndrome. Sci Rep. 2016; 6:34232.
Article93. Konstantinidou AE, Fryssira H, Sifakis S, Karadimas C, Kaminopetros P, Agrogiannis G, et al. Cranioectodermal dysplasia: a probable ciliopathy. Am J Med Genet A. 2009; 149A:2206–11.
Article94. Levin LS, Perrin JC, Ose L, Dorst JP, Miller JD, McKusick VA. A heritable syndrome of craniosynostosis, short thin hair, dental abnormalities, and short limbs: cranioectodermal dysplasia. J Pediatr. 1977; 90:55–61.
Article95. Fry AE, Klingenberg C, Matthes J, Heimdal K, Hennekam RC, Pilz DT. Connective tissue involvement in two patients with features of cranioectodermal dysplasia. Am J Med Genet A. 2009; 149A:2212–5.
Article96. Gilissen C, Arts HH, Hoischen A, Spruijt L, Mans DA, Arts P, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010; 87:418–23.
Article97. Walczak-Sztulpa J, Eggenschwiler J, Osborn D, Brown DA, Emma F, Klingenberg C, et al. Cranioectodermal dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet. 2010; 86:949–56.
Article98. Huber C, Wu S, Kim AS, Sigaudy S, Sarukhanov A, Serre V, et al. WDR34 mutations that cause short-rib polydactyly syndrome type III/severe asphyxiating thoracic dysplasia reveal a role for the NF-κB pathway in cilia. Am J Hum Genet. 2013; 93:926–31.
Article99. McInerney-Leo AM, Schmidts M, Cortes CR, Leo PJ, Gener B, Courtney AD, et al. Short-rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet. 2013; 93:515–23.
Article100. Tsurumi Y, Hamada Y, Katoh Y, Nakayama K. Interactions of the dynein-2 intermediate chain WDR34 with the light chains are required for ciliary retrograde protein trafficking. Mol Biol Cell. 2019; 30:658–70.
Article101. Asante D, Stevenson NL, Stephens DJ. Subunit composition of the human cytoplasmic dynein-2 complex. J Cell Sci. 2014; 127(Pt 21):4774–87.
Article102. Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. 2018; 39:152–66.
Article103. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004; 75:979–87.
Article104. Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013; 12:894–905.
Article105. Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet. 2009; 151C:326–40.
Article106. Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004; 125A:125–34.
Article107. Rivero O, Reif A, Sanjuan J, Molto MD, Kittel-Schneider S, Najera C, et al. Impact of the AHI1 gene on the vulnerability to schizophrenia: a case-control association study. PLoS One. 2010; 5:e12254.
Article108. Alvarez Retuerto AI, Cantor RM, Gleeson JG, Ustaszewska A, Schackwitz WS, Pennacchio LA, et al. Association of common variants in the Joubert syndrome gene (AHI1) with autism. Hum Mol Genet. 2008; 17:3887–96.
Article109. Weng L, Lin YF, Li AL, Wang CE, Yan S, Sun M, et al. Loss of Ahi1 affects early development by impairing BM88/Cend1-mediated neuronal differentiation. J Neurosci. 2013; 33:8172–84.
Article110. Xu X, Yang H, Lin YF, Li X, Cape A, Ressler KJ, et al. Neuronal Abelson helper integration site-1 (Ahi1) deficiency in mice alters TrkB signaling with a depressive phenotype. Proc Natl Acad Sci U S A. 2010; 107:19126–31.
Article111. Hilgendorf KI, Johnson CT, Jackson PK. The primary cilium as a cellular receiver: organizing ciliary GPCR signaling. Curr Opin Cell Biol. 2016; 39:84–92.
Article112. Koemeter-Cox AI, Sherwood TW, Green JA, Steiner RA, Berbari NF, Yoder BK, et al. Primary cilia enhance kisspeptin receptor signaling on gonadotropin-releasing hormone neurons. Proc Natl Acad Sci U S A. 2014; 111:10335–40.
Article113. Leaf A, Von Zastrow M. Dopamine receptors reveal an essential role of IFT-B, KIF17, and Rab23 in delivering specific receptors to primary cilia. Elife. 2015; 4:e06996.
Article114. Brailov I, Bancila M, Brisorgueil MJ, Miquel MC, Hamon M, Verge D. Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res. 2000; 872:271–5.
Article115. Higginbotham H, Guo J, Yokota Y, Umberger NL, Su CY, Li J, et al. Arl13b-regulated cilia activities are essential for polarized radial glial scaffold formation. Nat Neurosci. 2013; 16:1000–7.
Article116. Nauli SM, Jin X, Hierck BP. The mechanosensory role of primary cilia in vascular hypertension. Int J Vasc Med. 2011; 2011:376281.
Article117. McCormack SE, Li D, Kim YJ, Lee JY, Kim SH, Rapaport R, et al. Digenic inheritance of PROKR2 and WDR11 mutations in pituitary stalk interruption syndrome. J Clin Endocrinol Metab. 2017; 102:2501–7.
Article118. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am J Hum Genet. 2010; 87:465–79.
Article119. Izumi Y, Suzuki E, Kanzaki S, Yatsuga S, Kinjo S, Igarashi M, et al. Genome-wide copy number analysis and systematic mutation screening in 58 patients with hypogonadotropic hypogonadism. Fertil Steril. 2014; 102:1130–6.
Article120. Raivio T, Avbelj M, McCabe MJ, Romero CJ, Dwyer AA, Tommiska J, et al. Genetic overlap in Kallmann syndrome, combined pituitary hormone deficiency, and septo-optic dysplasia. J Clin Endocrinol Metab. 2012; 97:E694–9.
Article121. Kim SH. Congenital hypogonadotropic hypogonadism and Kallmann syndrome: past, present, and future. Endocrinol Metab (Seoul). 2015; 30:456–66.
Article122. Fraietta R, Zylberstejn DS, Esteves SC. Hypogonadotropic hypogonadism revisited. Clinics (Sao Paulo). 2013; 68 Suppl 1(Suppl 1):81–8.
Article123. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism. Pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2015; 11:547–64.
Article124. Bonomi M, Libri DV, Guizzardi F, Guarducci E, Maiolo E, Pignatti E, et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl. 2012; 14:49–56.
Article125. Brioude F, Bouligand J, Trabado S, Francou B, Salenave S, Kamenicky P, et al. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotypephenotype relationships. Eur J Endocrinol. 2010; 162:835–51.
Article126. Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, et al. WDR11-mediated Hedgehog signaling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. 2018; 19:269–89.
Article127. Wolf MT, Saunier S, O’Toole JF, Wanner N, Groshong T, Attanasio M, et al. Mutational analysis of the RPGRIP1L gene in patients with Joubert syndrome and nephronophthisis. Kidney Int. 2007; 72:1520–6.
Article128. Akcan N, Bas F, Poyrazoglu S, Bundak R. Joubert syndrome with multiple pituitary hormone deficiency. BMJ Case Rep. 2019; 12:e229016.
Article129. Niceta M, Dentici ML, Ciolfi A, Marini R, Barresi S, Lepri FR, et al. Co-occurrence of mutations in KIF7 and KIAA0556 in Joubert syndrome with ocular coloboma, pituitary malformation and growth hormone deficiency: a case report and literature review. BMC Pediatr. 2020; 20:120.
Article130. Li HL, Zhang YJ, Chen XP, Luo JQ, Liu SY, Zhang ZL. Association between GNB3 c.825C > T polymorphism and the risk of overweight and obesity: a meta-analysis. Meta Gene. 2016; 9:18–25.
Article131. Rizvi S, Raza ST, Rahman Q, Mahdi F. Role of GNB3, NET, KCNJ11, TCF7L2 and GRL genes single nucleotide polymorphism in the risk prediction of type 2 diabetes mellitus. 3 Biotech. 2016; 6:255.
Article132. Klenke S, Kussmann M, Siffert W. The GNB3 C825T polymorphism as a pharmacogenetic marker in the treatment of hypertension, obesity, and depression. Pharmacogenet Genomics. 2011; 21:594–606.
Article133. Comuzzie AG, Cole SA, Laston SL, Voruganti VS, Haack K, Gibbs RA, et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One. 2012; 7:e51954.
Article134. Green JS, Parfrey PS, Harnett JD, Farid NR, Cramer BC, Johnson G, et al. The cardinal manifestations of Bardet-Biedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med. 1989; 321:1002–9.
Article135. Adam MP, Ardinger HH, Pagon RA, Wallace SE. GeneReviews. Seattle: University of Washington, Seattle;1993-2020. Chapter, Bardet-Biedl syndrome [cited 2020 Jul 28]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1363.136. Kim SK, Shindo A, Park TJ, Oh EC, Ghosh S, Gray RS, et al. Planar cell polarity acts through septins to control collective cell movement and ciliogenesis. Science. 2010; 329:1337–40.
Article137. Toriyama M, Lee C, Taylor SP, Duran I, Cohn DH, Bruel AL, et al. The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat Genet. 2016; 48:648–56.
Article138. Heinen CA, Losekoot M, Sun Y, Watson PJ, Fairall L, Joustra SD, et al. Mutations in TBL1X are associated with central hypothyroidism. J Clin Endocrinol Metab. 2016; 101:4564–73.139. Sugisawa C, Takamizawa T, Abe K, Hasegawa T, Shiga K, Sugawara H, et al. Genetics of congenital isolated TSH deficiency: mutation screening of the known causative genes and a literature review. J Clin Endocrinol Metab. 2019; 104:6229–37.
Article140. Zwaveling-Soonawala N, Naafs JC, Verkerk PH, van Trotsenburg ASP. Mortality in children with early-detected congenital central hypothyroidism. J Clin Endocrinol Metab. 2018; 103:3078–82.
Article141. Brooks BP, Kleta R, Caruso RC, Stuart C, Ludlow J, Stratakis CA. Triple-A syndrome with prominent ophthalmic features and a novel mutation in the AAAS gene: a case report. BMC Ophthalmol. 2004; 4:7.142. Clark AJ, Weber A. Adrenocorticotropin insensitivity syndromes. Endocr Rev. 1998; 19:828–43.
Article143. Roucher-Boulez F, Brac de la Perriere A, Jacquez A, Chau D, Guignat L, Vial C, et al. Triple-A syndrome: a wide spectrum of adrenal dysfunction. Eur J Endocrinol. 2018; 178:199–207.
Article144. Tata B, Huijbregts L, Jacquier S, Csaba Z, Genin E, Meyer V, et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 2014; 12:e1001952.
Article145. Gobe C, Elzaiat M, Meunier N, Andre M, Sellem E, Congar P, et al. Dual role of DMXL2 in olfactory information transmission and the first wave of spermatogenesis. PLoS Genet. 2019; 15:e1007909.
Article
