J Clin Neurol.
2019 Apr;15(2):184-190. 10.3988/jcn.2019.15.2.184.
Analysis of 12 Chinese Patients with Proline-to-Leucine Mutation at Codon 102-Associated Gerstmann-Sträussler-Scheinker Disease
- Affiliations
-
- 1State Key Laboratory for Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases (Zhejiang University), National Institute for Viral Disease Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China. dongxp238@sina.com
- 2Center of Global Public Health, Chinese Center for Disease Control and Prevention, Beijing, China.
- KMID: 2467732
- DOI: http://doi.org/10.3988/jcn.2019.15.2.184
Abstract
- BACKGROUND AND PURPOSE
Gerstmann-Sträussler-Scheinker disease (GSS) with a proline-to-leucine mutation at codon 102 (P102L) in the PRNP gene is the most frequently reported GSS subtype worldwide. This study aimed to determine the epidemiological, clinical, genetic, and laboratory characteristics of 12 Chinese patients with P102L-associated GSS (henceforth P102L GSS).
METHODS
The enrolled P102L GSS cases were analyzed according to the diagnostic criteria for Creutzfeldt-Jakob disease (CJD) issued by the China National Health Commission.
RESULTS
The median onset age was 50 years (range 34 to 67 years) and sex ratio was 1:2 (males:females). Most patients displayed more than one foremost symptom. Movement symptoms were frequently reported (9 of the 12 cases), followed by rapidly progressing dementia (7 cases), mental problems (5 cases), and slowly progressing dementia (2 cases). Almost all cases displayed more sporadic CJD (sCJD)-associated neurological symptoms and signs as time progressed. Five (45.5%) of 11 cases were cerebrospinal fluid 14-3-3 positive, and 2 (25%) of 8 cases exhibited periodic sharp wave complexes in electroencephalograms. MRI abnormalities were detected in all 11 of the scanned patients. Methionine homozygous genotype at codon 129 (M129M) and glutamic acid homozygous at codon 219 (E219E) homozygosity was present in 11 cases, while 1 case was M129M homozygous and glutamic acid/lysine heterozygous at codon 219 (E219K) heterozygous. Ten of the 12 cases recalled a disease-related family history during the clinical interviews. The median survival from symptom onset of the seven dead cases was 16 months (range 10 to 44 months). Patients showing the sCJD phenotype (rapidly progressing dementia) appeared to be associated with a shorter survival time.
CONCLUSIONS
The indistinguishable clinical features of P102L GSS patients with sCJD, especially in the early stage, support the importance of PRNP testing for diagnosing GSS.
Keyword
MeSH Terms
Figure
-

Fig. 1 Distribution of the onset ages of 12 Chinese P102L Gerstmann-Sträussler-Scheinker disease patients. The median onset ages overall and in the different groups are shown at the top right.

Fig. 2 Survival times of 12 P102L Gerstmann-Sträussler-Scheinker disease patients according to gender (males and females, A) and onset age (<50 years or ≥50 years, B).
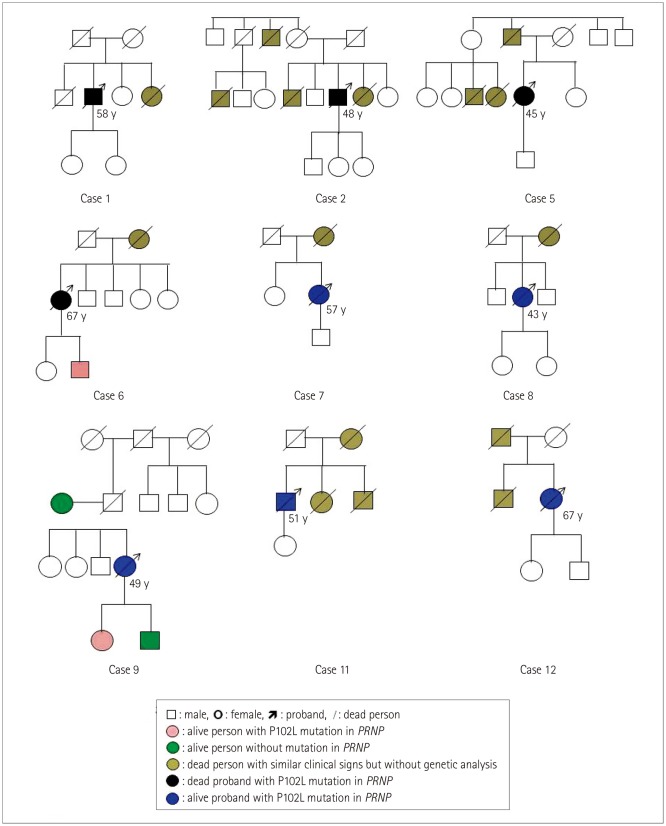
Fig. 3 Disease-related family histories and P102L mutations in PRNP in several families. The shapes, lines, and colors are defined in the panel below the figure. P102L: proline-to-leucine mutation at codon 102.
Reference
-
1. Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998; 95:13363–13383. PMID: 9811807.2. Takada LT, Kim MO, Metcalf S, Gala II, Geschwind MD. Prion disease. Handb Clin Neurol. 2018; 148:441–464. PMID: 29478593.
Article3. Kim MO, Takada LT, Wong K, Forner SA, Geschwind MD. Genetic PrP prion diseases. Cold Spring Harb Perspect Biol. 2018; 10:a033134. PMID: 28778873.
Article4. Hainfellner JA, Brantner-Inthaler S, Cervenáková L, Brown P, Kitamoto T, Tateishi J, et al. The original Gerstmann-Sträussler-Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol. 1995; 5:201–211. PMID: 8520719.
Article5. Lloyd S, Mead S, Collinge J. Genetics of prion disease. Top Curr Chem. 2011; 305:1–22. PMID: 21528440.
Article6. Doh-ura K, Tateishi J, Sasaki H, Kitamoto T, Sakaki Y. Pro→Leu change at position 102 of prion protein is the most common but not the sole mutation related to Gerstmann-Sträussler syndrome. Biochem Biophys Res Commun. 1989; 163:974–979. PMID: 2783132.7. Minikel EV, Vallabh SM, Lek M, Estrada K, Samocha KE, Sathirapongsasuti JF, et al. Quantifying prion disease penetrance using large population control cohorts. Sci Transl Med. 2016; 8:322ra9.
Article8. Hsiao K, Dlouhy SR, Farlow MR, Cass C, Da Costa M, Conneally PM, et al. Mutant prion proteins in Gerstmann-Sträussler-Scheinker disease with neurofibrillary tangles. Nat Genet. 1992; 1:68–71. PMID: 1363810.
Article9. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD, et al. Linkage of a prion protein missense variant to Gerstmann-Sträussler syndrome. Nature. 1989; 338:342–345. PMID: 2564168.
Article10. Takada LT, Kim MO, Cleveland RW, Wong K, Forner SA, Gala II, et al. Genetic prion disease: experience of a rapidly progressive dementia center in the United States and a review of the literature. Am J Med Genet B Neuropsychiatr Genet. 2017; 174:36–69. PMID: 27943639.
Article11. Shi Q, Zhou W, Chen C, Zhang BY, Xiao K, Zhang XC, et al. The features of genetic prion diseases based on Chinese surveillance program. PLoS One. 2015; 10:e0139552. PMID: 26488179.
Article12. Aguzzi A. Prion diseases of humans and farm animals: epidemiology, genetics, and pathogenesis. J Neurochem. 2006; 97:1726–1739. PMID: 16805779.
Article13. Unterberger U, Voigtländer T, Budka H. Pathogenesis of prion diseases. Acta Neuropathol. 2005; 109:32–48. PMID: 15645262.
Article14. Chen C, Dong XP. Epidemiological characteristics of human prion diseases. Infect Dis Poverty. 2016; 5:47. PMID: 27251305.
Article15. Webb TE, Poulter M, Beck J, Uphill J, Adamson G, Campbell T, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008; 131:2632–2646. PMID: 18757886.
Article16. Krasnianski A, Heinemann U, Ponto C, Kortt J, Kallenberg K, Varges D, et al. Clinical findings and diagnosis in genetic prion diseases in Germany. Eur J Epidemiol. 2016; 31:187–196. PMID: 26076917.
Article17. Higuma M, Sanjo N, Satoh K, Shiga Y, Sakai K, Nozaki I, et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PLoS One. 2013; 8:e60003. PMID: 23555862.
Article18. Gao C, Shi Q, Tian C, Chen C, Han J, Zhou W, et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PLoS One. 2011; 6:e24231. PMID: 21904617.
Article19. Zerr I, Kallenberg K, Summers DM, Romero C, Taratuto A, Heinemann U, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain. 2009; 132:2659–2668. PMID: 19773352.
Article20. Shi Q, Zhou W, Chen C, Gao C, Xiao K, Wang J, et al. Quality evaluation for the surveillance system of human prion diseases in China based on the data from 2010 to 2016. Prion. 2016; 10:484–491. PMID: 27690734.
Article21. Geschwind MD. Prion diseases. Continuum (Minneap Minn). 2015; 21:1612–1638. PMID: 26633779.
Article22. Vitali P, Maccagnano E, Caverzasi E, Henry RG, Haman A, Torres-Chae C, et al. Diffusion-weighted MRI hyperintensity patterns differentiate CJD from other rapid dementias. Neurology. 2011; 76:1711–1719. PMID: 21471469.
Article23. Kovács GG, Puopolo M, Ladogana A, Pocchiari M, Budka H, Van Duijn C, et al. Genetic prion disease: the EUROCJD experience. Hum Genet. 2005; 118:166–174. PMID: 16187142.
Article24. Mead S, Uphill J, Beck J, Poulter M, Campbell T, Lowe J, et al. Genome-wide association study in multiple human prion diseases suggests genetic risk factors additional to PRNP. Hum Mol Genet. 2012; 21:1897–1906. PMID: 22210626.
Article25. Lloyd SE, Mead S, Collinge J. Genetics of prion diseases. Curr Opin Genet Dev. 2013; 23:345–351. PMID: 23518043.
Article26. Jeong BH, Nam JH, Lee YJ, Lee KH, Jang MK, Carp RI, et al. Polymorphisms of the prion protein gene (PRNP) in a Korean population. J Hum Genet. 2004; 49:319–324. PMID: 15148589.
Article27. Bishop MT, Pennington C, Heath CA, Will RG, Knight RS. PRNP variation in UK sporadic and variant Creutzfeldt Jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med Genet. 2009; 10:146. PMID: 20035629.
Article28. Tanaka Y, Minematsu K, Moriyasu H, Yamaguchi T, Yutani C, Kitamoto T, et al. A Japanese family with a variant of Gerstmann-Sträussler-Scheinker disease. J Neurol Neurosurg Psychiatry. 1997; 62:454–457. PMID: 9153600.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Gerstmann-Sträussler-Scheinker Disease (Pro102Leu) Presenting as Rapidly Progressive Dementia
- A Case of Gerstmann-Straussler-Scheinker Disease
- Accumulation Area of a Japanese PRNP P102L Variant Associated With Gerstmann–Sträussler–Scheinker Disease: The Ariake PRNP P102L Variant
- Gerstmann-SträusslerScheinker Disease: A Case Report
- Possible Role of a Missense Mutation of p.P167S on NOTCH3 Gene Associated with Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
