Electrophysiological characteristics of R47W and A298T mutations in CLC-1 of myotonia congenita patients and evaluation of clinical features
- Affiliations
-
- 1Department of Physiology, Seoul National University College of Medicine, Seoul 03080, Korea. insuk@snu.ac.kr
- 2Department of Neurology, Research Institute for Convergence of Biomedical Research and Technology, Pusan University Yangsan Hospital, Yangsan 50612, Korea. dskim@pusan.ac.kr
- KMID: 2384458
- DOI: http://doi.org/10.4196/kjpp.2017.21.4.439
Abstract
- Myotonia congenita (MC) is a genetic disease that displays impaired relaxation of skeletal muscle and muscle hypertrophy. This disease is mainly caused by mutations of CLCN1 that encodes human skeletal muscle chloride channel (CLC-1). CLC-1 is a voltage gated chloride channel that activates upon depolarizing potentials and play a major role in stabilization of resting membrane potentials in skeletal muscle. In this study, we report 4 unrelated Korean patients diagnosed with myotonia congenita and their clinical features. Sequence analysis of all coding regions of the patients was performed and mutation, R47W and A298T, was commonly identified. The patients commonly displayed transient muscle weakness and only one patient was diagnosed with autosomal dominant type of myotonia congenita. To investigate the pathological role of the mutation, electrophysiological analysis was also performed in HEK 293 cells transiently expressing homo- or heterodimeric mutant channels. The mutant channels displayed reduced chloride current density and altered channel gating. However, the effect of A298T on channel gating was reduced with the presence of R47W in the same allele. This analysis suggests that impaired CLC-1 channel function can cause myotonia congenita and that R47W has a protective effect on A298T in relation to channel gating. Our results provide clinical features of Korean myotonia congenita patients who have the heterozygous mutation and reveal underlying pathophyological consequences of the mutants by taking electrophysiological approach.
Keyword
MeSH Terms
Figure
-
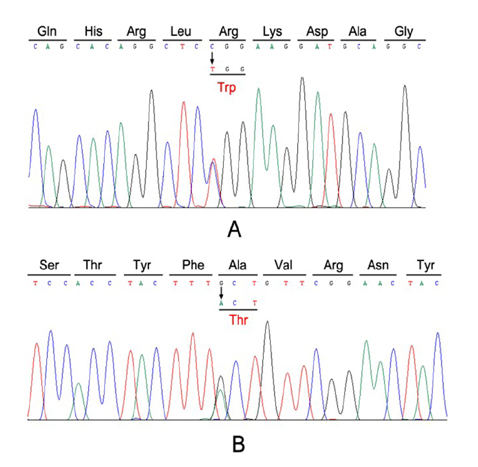
Fig. 1 Recurrent mutation, c139C>T and c.892G>A, of hCLCN1 found in myotonia congenita patients. (A) C to C/T at position c.139, results in a substitution of amino acid tryptophan (TGG) for arginine (CGG) at position p.47 (p.Arg47Trp). (B) G to G/A at position c.892, results in a substitution of amino acid threonin (ACT) for alanine (GCT) at position p.298 (p.Ala298Thr).
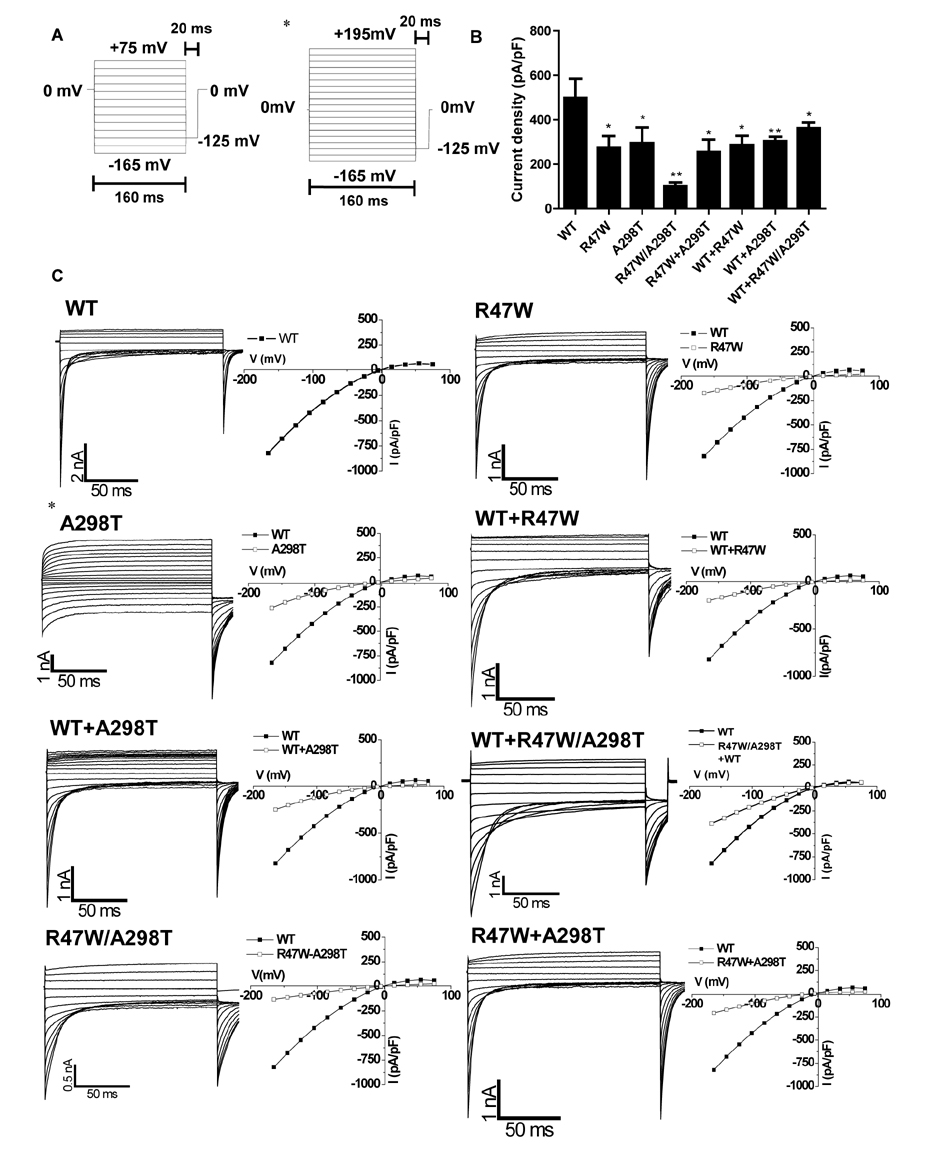
Fig. 2 The representative chloride currents recorded from HEK293 cells transiently expressing hCLC-1 WT channels and the myotonic mutants. (A) Voltage pulse protocol applied to the whole cell configuration. The voltage pulses were applied from −165 mV to +75 mV or +195 mV in 20 mV increments. A tail pulse was given at −125 mV during 20 ms and holding potential was at 0 mV. (B) The chloride current density. The maximal inwardly rectifying currents (pA) were obtained and divided by capacitance (pF). The values were recorded in absolute value. Data are means±s.e.m. n=10 (WT CLC-1), n=4 (R47W), n=4 (A298T), n=4 (WT+R47W), n=5 (WT+A298T), n=4 (WT+R47W/A298T), n=4 (R47W/A298T), n=5 (R47W+A298T). Statistical significance (*p<0.05, **p<0.01) was determined by student's t-test. (C) The kinetics and I (current)-V (voltage) relationship of heteromeric and homodimeric R47W. (Left) Current traces of WT hCLC-1 channel and the heteromeric or homodimeric mutants obtained by whole cell patch clamp. (Right) Comparison of IV relationship in WT and the heteromeric or homodimeric mutants. The maximal inwardly rectifying currents (pA) were divided by the whole cell capacitance (pF). Asterisk indicates the mutant recorded by the voltage pulse protocol to +195 mV. Closed and open square indicates WT hCLC-1 and the mutants, respectively.

Fig. 3 Voltage dependence of the relative open probability obtained from WT hCLC-1 and the myotonic mutants. (A) The analysis of voltage dependence of the WT hCLC-1, homo-, or heterodimeric mutants. (Left) Illustration of tail currents from 150 ms to 170 ms. (Right) The comparison of open probability (Po) of the WT hCLC-1, homo-, or heterodimeric mutants. The peak currents at tail pulse were divided by the minimal current, and fitted to Boltzmann distributions. Black and red line indicate the WT hCLC-1, homo-, or heterodimeric mutants, respectively. (B) The comparison of voltage dependences obtained from WT hCLC-1 and the mutants. Data are means±s.e.m. n=10 (WT CLC-1), n=4 (R47W), n=4 (A298T), n=4 (WT+R47W), n=5 (WT + A298T), n=4 (WT+R47W/A298T), n=4 (R47W/A298T), n=5 (R47W+A298T).
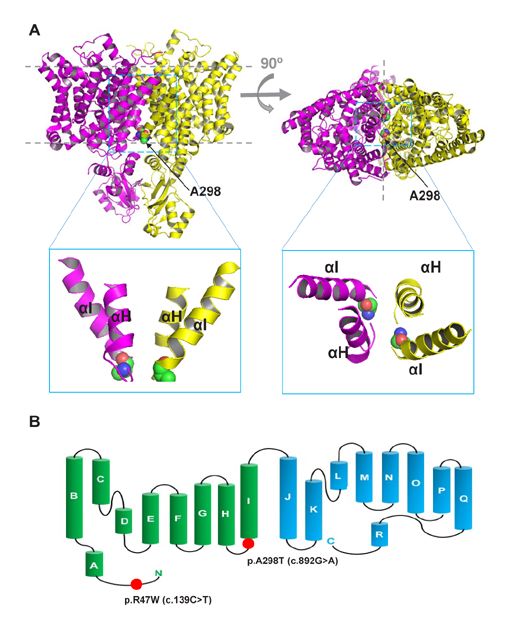
Fig. 4 Location of the mutation in homology modeling of hCLC-1 based on the crystal structure of cmCLC and CLC-K. (A) Side (left) and top (right) view of homology modeling describing the homodimeric structure of hCLC-1 channel. The structure was built based on the structure of stCLC (PDB id: 1KPL) and CLC-K (PDB id: 5TQQ). Each monomer was colored in magenta and yellow and the location of mutant corresponding to A298T was indicated in the spacefilled model in cyan. The illustrations in the box depict the position of the A298T mutant localized on the loop between helix H and I. (B) Location of the mutants, R47W and A298T depicted on the topology model of hCLC-1. The model of hCLC-1 channel was described based on the structure from Dutzler et al. [13].
Reference
-
1. Koch MC, Steinmeyer K, Lorenz C, Ricker K, Wolf F, Otto M, Zoll B, Lehmann-Horn F, Grzeschik KH, Jentsch TJ. The skeletal muscle chloride channel in dominant and recessive human myotonia. Science. 1992; 257:797–800.2. George AL Jr, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC. Molecular basis of Thomsen's disease (autosomal dominant myotonia congenita). Nat Genet. 1993; 3:305–310.3. Wu FF, Ryan A, Devaney J, Warnstedt M, Korade-Mirnics Z, Poser B, Escriva MJ, Pegoraro E, Yee AS, Felice KJ, Giuliani MJ, Mayer RF, Mongini T, Palmucci L, Marino M, Rüdel R, Hoffman EP, Fahlke C. Novel CLCN1 mutations with unique clinical and electrophysiological consequences. Brain. 2002; 125:2392–2407.4. Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002; 82:503–568.5. Lorenz C, Meyer-Kleine C, Steinmeyer K, Koch MC, Jentsch TJ. Genomic organization of the human muscle chloride channel CIC-1 and analysis of novel mutations leading to Becker-type myotonia. Hum Mol Genet. 1994; 3:941–946.6. Liu XL, Huang XJ, Shen JY, Zhou HY, Luan XH, Wang T, Chen SD, Wang Y, Tang HD, Cao L. Myotonia congenita: novel mutations in CLCN1 gene. Channels (Austin). 2015; 9:292–298.7. Pedersen TH, Riisager A, de Paoli FV, Chen TY, Nielsen OB. Role of physiological ClC-1 Cl- ion channel regulation for the excitability and function of working skeletal muscle. J Gen Physiol. 2016; 147:291–308.8. Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature. 1991; 354:301–304.9. Miller C. ClC chloride channels viewed through a transporter lens. Nature. 2006; 440:484–489.10. Jentsch TJ. CLC chloride channels and transporters: from genes to protein structure, pathology and physiology. Crit Rev Biochem Mol Biol. 2008; 43:3–36.11. Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010; 330:635–641.12. Park E, Campbell EB, MacKinnon R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature. 2017; 541:500–505.13. Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002; 415:287–294.14. Accardi A, Pusch M. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J Gen Physiol. 2000; 116:433–444.15. Fahlke C. Ion permeation and selectivity in ClC-type chloride channels. Am J Physiol Renal Physiol. 2001; 280:F748–F757.16. Moon IS, Kim HS, Shin JH, Park YE, Park KH, Shin YB, Bae JS, Choi YC, Kim DS. Novel CLCN1 mutations and clinical features of Korean patients with myotonia congenita. J Korean Med Sci. 2009; 24:1038–1044.17. Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS. Clinical diversity of SCN4A-mutation-associated skeletal muscle sodium channelopathy. J Clin Neurol. 2009; 5:186–191.18. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993; 234:779–815.19. DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002; 12:14–20.20. Gao F, Ma FC, Yuan ZF, Yang CW, Li HF, Xia ZZ, Shui QX, Jiang KW. Novel chloride channel gene mutations in two unrelated Chinese families with myotonia congenita. Neurol India. 2010; 58:743–746.21. Fialho D, Schorge S, Pucovska U, Davies NP, Labrum R, Haworth A, Stanley E, Sud R, Wakeling W, Davis MB, Kullmann DM, Hanna MG. Chloride channel myotonia: exon 8 hot-spot for dominant-negative interactions. Brain. 2007; 130:3265–3274.22. Cederholm JM, Rychkov GY, Bagley CJ, Bretag AH. Inter-subunit communication and fast gate integrity are important for common gating in hClC-1. Int J Biochem Cell Biol. 2010; 42:1182–1188.23. Duffield M, Rychkov G, Bretag A, Roberts M. Involvement of helices at the dimer interface in ClC-1 common gating. J Gen Physiol. 2003; 121:149–161.24. Lee TT, Zhang XD, Chuang CC, Chen JJ, Chen YA, Chen SC, Chen TY, Tang CY. Myotonia congenita mutation enhances the degradation of human CLC-1 chloride channels. PLoS One. 2013; 8:e55930.
