Ann Lab Med.
2017 Jan;37(1):58-62. 10.3343/alm.2017.37.1.58.
Novel Pathogenic Variant (c.580C>T) in the CPS1 Gene in a Newborn With Carbamoyl Phosphate Synthetase 1 Deficiency Identified by Whole Exome Sequencing
- Affiliations
-
- 1Department of Laboratory Medicine and Genetics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. nayadoo@hanmail.net
- 2Department of Laboratory Medicine, Seoul National University College of Medicine, Seoul National University Bundang Hospital, Seongnam, Korea.
- 3Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea. ws123.park@samsung.com
- 4Samsung Biomedical Research Institute, Sungkyunkwan University School of Medicine, Seoul, Korea.
- KMID: 2373615
- DOI: http://doi.org/10.3343/alm.2017.37.1.58
Abstract
- Diagnosis of the urea cycle disorder (USD) carbamoyl-phosphate synthetase 1 (CPS1) deficiency (CPS1D) based on only the measurements of biochemical intermediary metabolites is not sufficient to properly exclude other UCDs with similar symptoms. We report the first Korean CPS1D patient using whole exome sequencing (WES). A four-day-old female neonate presented with respiratory failure due to severe metabolic encephalopathy with hyperammonemia (1,690 µmol/L; reference range, 11.2-48.2 µmol/L). Plasma amino acid analysis revealed markedly elevated levels of alanine (2,923 µmol/L; reference range, 131-710 µmol/L) and glutamine (5,777 µmol/L; reference range, 376-709 µmol/L), whereas that of citrulline was decreased (2 µmol/L; reference range, 10-45 µmol/L). WES revealed compound heterozygous pathogenic variants in the CPS1 gene: one novel nonsense pathogenic variant of c.580C>T (p.Gln194*) and one known pathogenic frameshift pathogenic variant of c.1547delG (p.Gly516Alafs*5), which was previously reported in Japanese patients with CPS1D. We successfully applied WES to molecularly diagnose the first Korean patient with CPS1D in a clinical setting. This result supports the clinical applicability of WES for cost-effective molecular diagnosis of UCDs.
Keyword
MeSH Terms
-
Base Sequence
Carbamoyl-Phosphate Synthase (Ammonia)/chemistry/*genetics
Carbamoyl-Phosphate Synthase I Deficiency Disease/diagnosis/*genetics
Codon, Nonsense
Exons
Female
Frameshift Mutation
High-Throughput Nucleotide Sequencing
Humans
Infant, Newborn
Republic of Korea
Sequence Analysis, DNA
Urea Cycle Disorders, Inborn/diagnosis
Carbamoyl-Phosphate Synthase (Ammonia)
Codon, Nonsense
Figure
-
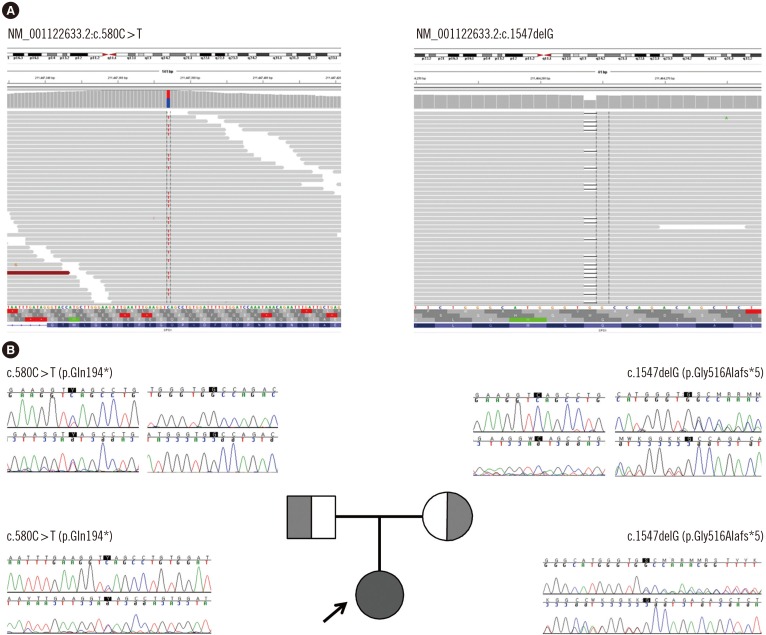
Fig. 1 CPS1 pathogenic variants identified in a patient with carbamoyl-phosphate synthetase 1 deficiency and her family members. CPS1 pathogenic variants were identified by whole exome sequencing in the patient (A) and were confirmed by Sanger sequencing in the patient and her family members (B).
Reference
-
1. Häberle J, Shchelochkov OA, Wang J, Katsonis P, Hall L, Reiss S, et al. Molecular defects in human carbamoy phosphate synthetase I: mutational spectrum, diagnostic and protein structure considerations. Hum Mutat. 2011; 32:579–589. PMID: 21120950.2. Ah Mew N, Lanpher BC, et al. Urea cycle disorders overview. In : Pagon RA, Adam MP, editors. GeneReviews(R). Seattle, WA: University of Washington;2011.3. Díez-Fernández C, Hu L, Cervera J, Häberle J, Rubio V. Understanding carbamoyl phosphate synthetase (CPS1) deficiency by using the recombinantly purified human enzyme: effects of CPS1 mutations that concentrate in a central domain of unknown function. Mol Genet Metab. 2014; 112:123–132. PMID: 24813853.4. Amstutz U, Andrey-Zürcher G, Suciu D, Jaggi R, Häberle J, Largiadèr CR. Sequence capture and next-generation resequencing of multiple tagged nucleic acid samples for mutation screening of urea cycle disorders. Clin Chem. 2011; 57:102–111. PMID: 21068339.5. Choi R, Woo HI, Choe BH, Park S, Yoon Y, Ki CS, et al. Application of whole exome sequencing to a rare inherited metabolic disease with neurological and gastrointestinal manifestations: a congenital disorder of glycosylation mimicking glycogen storage disease. Clin Chim Acta. 2015; 444:50–53. PMID: 25681648.6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17:405–424. PMID: 25741868.7. Wakutani Y, Nakayasu H, Takeshima T, Adachi M, Kawataki M, Kihira K, et al. Mutational analysis of carbamoylphosphate synthetase I deficiency in three Japanese patients. J Inherit Metab Dis. 2004; 27:787–788. PMID: 15617192.8. Kurokawa K, Yorifuji T, Kawai M, Momoi T, Nagasaka H, Takayanagi M, et al. Molecular and clinical analyses of Japanese patients with carbamoylphosphate synthetase 1 (CPS1) deficiency. J Hum Genet. 2007; 52:349–354. PMID: 17310273.9. Summar ML, Koelker S, Freedenberg D, Le Mons C, Haberle J, Lee HS, et al. The incidence of urea cycle disorders. Mol Genet Metab. 2013; 110:179–180. PMID: 23972786.10. Finckh U, Kohlschütter A, Schäfer H, Sperhake K, Colombo JP, Gal A. Prenatal diagnosis of carbamoyl phosphate synthetase I deficiency by identification of a missense mutation in CPS1. Hum Mutat. 1998; 12:206–211. PMID: 9711878.11. Aoshima T, Kajita M, Sekido Y, Mimura S, Itakura A, Yasuda I, et al. Carbamoyl phosphate synthetase I deficiency: molecular genetic findings and prenatal diagnosis. Prenat Diagn. 2001; 21:634–637. PMID: 11536261.12. Carss KJ, Hillman SC, Parthiban V, McMullan DJ, Maher ER, Kilby MD, et al. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet. 2014; 23:3269–3277. PMID: 24476948.13. Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014; 370:2418–2425. PMID: 24941179.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Whole-exome sequencing analysis in a case of primary congenital glaucoma due to the partial uniparental isodisomy
- A Familial Case with Phenotypic Differences in a CAV3 Pathogenic Variant
- Novel Pathogenic Variant (c.3178G>A) in the SMC1A Gene in a Family With Cornelia de Lange Syndrome Identified by Exome Sequencing
- The First Korean Case of Gorlin?Goltz Syndrome Caused by a PTCH2 Pathogenic Variant Identified via Whole Exome Sequencing
- Identification of a De Novo Heterozygous Missense FLNB Mutation in Lethal Atelosteogenesis Type I by Exome Sequencing
