Translational Read-Through of a Nonsense Mutation Causing Bartter Syndrome
- Affiliations
-
- 1Department of Pediatrics, Samsung Medical Center, Sungkyunkwan University School of Medicine, Seoul, Korea.
- 2Department of Pediatrics, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Korea.
- 3Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea. cheonghi@snu.ac.kr
- 4Research Coordination Center for Rare Diseases, Seoul National University Hospital, Seoul, Korea.
- 5Kidney Research Institute, Medical Research Center, Seoul National University College of Medicine, Seoul, Korea.
- KMID: 2158027
- DOI: http://doi.org/10.3346/jkms.2013.28.6.821
Abstract
- Bartter syndrome (BS) is classified into 5 genotypes according to underlying mutant genes and BS III is caused by loss-of-function mutations in the CLCNKB gene encoding for basolateral ClC-Kb. BS III is the most common genotype in Korean patients with BS and W610X is the most common CLCNKB mutation in Korean BS III. In this study, we tested the hypothesis that the CLCNKB W610X mutation can be rescued in vitro using aminoglycoside antibiotics, which are known to induce translational read-through of a nonsense mutation. The CLCNKB cDNA was cloned into a eukaryotic expression vector and the W610X nonsense mutation was generated by site-directed mutagenesis. Cultured polarized MDCK cells were transfected with the vectors, and the read-through was induced using an aminoglycoside derivative, G418. Cellular expression of the target protein was monitored via immunohistochemistry. While cells transfected with the mutant CLCNKB failed to express ClC-Kb, G418 treatment of the cells induced the full-length protein expression, which was localized to the basolateral plasma membranes. It is demonstrated that the W610X mutation in CLCNKB can be a good candidate for trial of translational read-through induction as a therapeutic modality.
MeSH Terms
-
Animals
Bartter Syndrome/genetics/*pathology
Chloride Channels/analysis/genetics/*metabolism
Cloning, Molecular
Codon, Nonsense
Dogs
Humans
Immunohistochemistry
Madin Darby Canine Kidney Cells
Microscopy, Confocal
Mutagenesis, Site-Directed
Recombinant Fusion Proteins/analysis/biosynthesis/genetics
Transfection
Chloride Channels
Codon, Nonsense
Recombinant Fusion Proteins
Figure
-
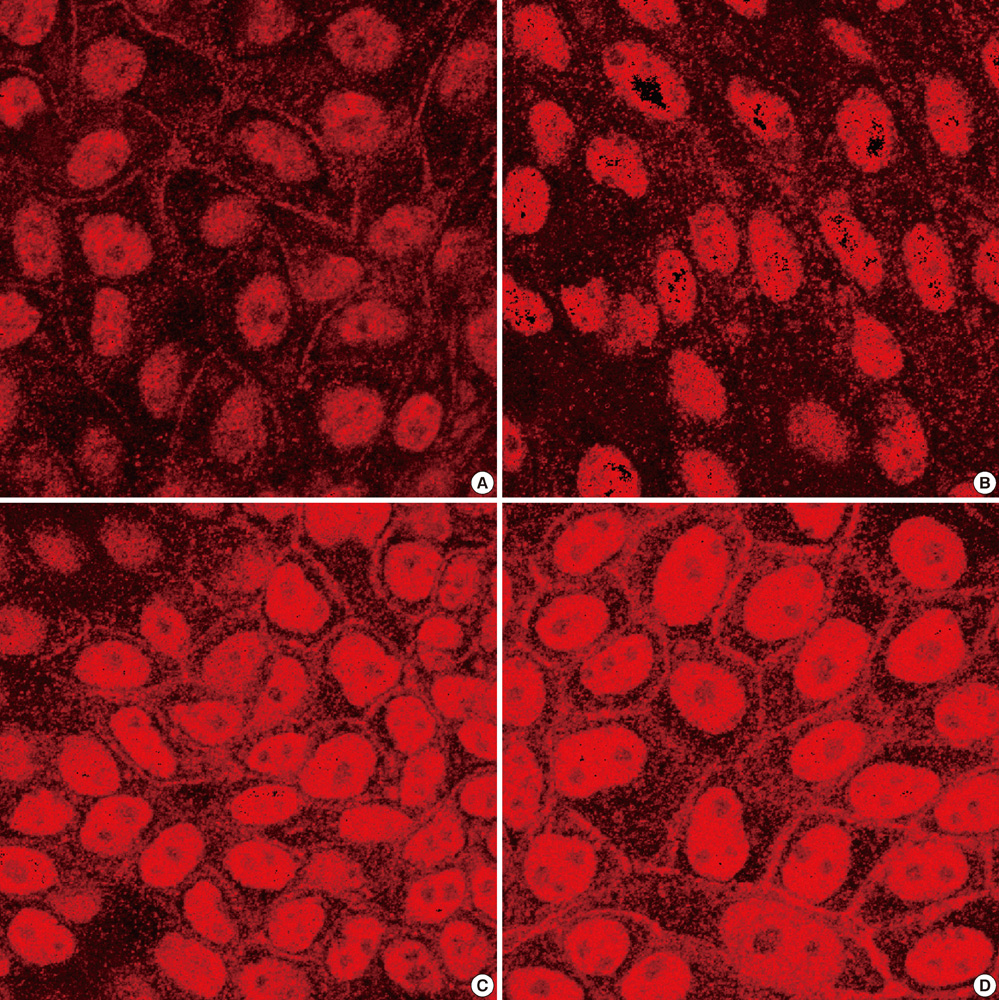
Fig. 1 Immunohistochemistry. Expression pattern of ClC-Kb-V5-His protein in polarized MDCK was observed after staining with C-terminal His antibody. While cells transfected with wild-type CLCNKB express ClC-Kb-V5-His along the plasma membranes (A), cells transfected with W610X mutant CLCNKB do not express the protein (B). After G418 treatment, cells transfected with mutant CLCNKB express the protein as same pattern as cells transfected with wild-type CLCNKB (C, D). The degree of expression shows no difference between cells treated with 75 µg/mL concentration of G418 (C) and 150 µg/mL concentration of G418 (D). All are in × 800 magnification.

Fig. 2 Confocal laser microscopic examination. Expression pattern of ClC-Kb-V5-His protein in polarized MDCK was observed after staining with C-terminal His antibody. In cells transfected with mutant CLCNKB, G418 treatment induces expression of ClC-Kb-V5-His protein along the plasma membrane. The degree of expression shows no difference between cells treated with 75 µg/mL concentration of G418 (A) and 150 µg/mL concentration of G418 (B) (Red is His and blue is DAPI). Arrows indicate the protein stained basolaterally and the expression is localized to the basolateral membranes but not in the apical membranes (B). Both are in × 800 magnification.
Reference
-
1. Schwarz I, Alon U. Bartter syndrome revisited. J Nephrol. 1996; 9:81–87.2. Bartter FC, Pronove P, Gill JR Jr, MacCardle RC. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am J Med. 1962; 33:811–828.3. Simon DB, Karet FE, Hamdan JM, DiPietro A, Sanjad SA, Lifton RP. Bartter's syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat Genet. 1996; 13:183–188.4. International Collaborative Study Group for Bartter-like Syndromes. Mutations in the gene encoding the inwardly-rectifying renal potassium channel, ROMK, cause the antenatal variant of Bartter syndrome: evidence for genetic heterogeneity. Hum Mol Genet. 1997; 6:17–26.5. Konrad M, Vollmer M, Lemmink HH, van den Heuvel LP, Jeck N, Vargas-Poussou R, Lakings A, Ruf R, Deschênes G, Antignac C, et al. Mutations in the chloride channel gene CLCNKB as a cause of classic Bartter syndrome. J Am Soc Nephrol. 2000; 11:1449–1459.6. Waldegger S, Jentsch TJ. Functional and structural analysis of ClC-K chloride channels involved in renal disease. J Biol Chem. 2000; 275:24527–24533.7. Birkenhäger R, Otto E, Schürmann MJ, Vollmer M, Ruf EM, Maier-Lutz I, Beekmann F, Fekete A, Omran H, Feldmann D, et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat Genet. 2001; 29:310–314.8. Watanabe S, Fukumoto S, Chang H, Takeuchi Y, Hasegawa Y, Okazaki R, Chikatsu N, Fujita T. Association between activating mutations of calcium-sensing receptor and Bartter's syndrome. Lancet. 2002; 360:692–694.9. Lee BH, Cho HY, Lee H, Han KH, Kang HG, Ha IS, Lee JH, Park YS, Shin JI, Lee DY, et al. Genetic basis of Bartter syndrome in Korea. Nephrol Dial Transplant. 2012; 27:1516–1521.10. Kellermayer R. Translational readthrough induction of pathogenic nonsense mutations. Eur J Med Genet. 2006; 49:445–450.11. Azimov R, Abuladze N, Sassani P, Newman D, Kao L, Liu W, Orozco N, Ruchala P, Pushkin A, Kurtz I. G418-mediated ribosomal read-through of a nonsense mutation causing autosomal recessive proximal renal tubular acidosis. Am J Physiol Renal Physiol. 2008; 295:F633–F641.12. Sangkuhl K, Schulz A, Römpler H, Yun J, Wess J, Schöneberg T. Aminoglycoside-mediated rescue of a disease-causing nonsense mutation in the V2 vasopressin receptor gene in vitro and in vivo. Hum Mol Genet. 2004; 13:893–903.13. Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, Sorscher EJ, Clancy JP. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007; 37:347–356.14. Lai CH, Chun HH, Nahas SA, Mitui M, Gamo KM, Du L, Gatti RA. Correction of ATM gene function by aminoglycoside-induced read-through of premature termination codons. Proc Natl Acad Sci U S A. 2004; 101:15676–15681.15. James PD, Raut S, Rivard GE, Poon MC, Warner M, McKenna S, Leggo J, Lillicrap D. Aminoglycoside suppression of nonsense mutations in severe hemophilia. Blood. 2005; 106:3043–3048.16. Popescu AC, Sidorova E, Zhang G, Eubanks JH. Aminoglycoside-mediated partial suppression of MECP2 nonsense mutations responsible for Rett syndrome in vitro. J Neurosci Res. 2010; 88:2316–2324.17. Yang C, Feng J, Song W, Wang J, Tsai B, Zhang Y, Scaringe WA, Hill KA, Margaritis P, High KA, et al. A mouse model for nonsense mutation bypass therapy shows a dramatic multiday response to geneticin. Proc Natl Acad Sci U S A. 2007; 104:15394–15399.18. Hyde SC, Gill DR. Ignoring the nonsense: a phase II trial in cystic fibrosis. Lancet. 2008; 372:691–692.19. Moosajee M, Gregory-Evans K, Ellis CD, Seabra MC, Gregory-Evans CY. Translational bypass of nonsense mutations in zebrafish rep1, pax2.1 and lamb1 highlights a viable therapeutic option for untreatable genetic eye disease. Hum Mol Genet. 2008; 17:3987–4000.20. Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003; 349:1433–1441.21. Nilsson M, Rydén-Aulin M. Glutamine is incorporated at the nonsense codons UAG and UAA in a suppressor-free Escherichia coli strain. Biochim Biophys Acta. 2003; 1627:1–6.22. Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop condon readthrough: potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000; 48:164–169.23. Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000; 6:1044–1055.24. Kaufman RJ. Correction of genetic disease by making sense from nonsense. J Clin Invest. 1999; 104:367–368.25. Maldague P, Laurent G, Carlier MB, Tulkens P. A 2'guanidyl derivative of gentamicin (S86451) with reduced nephrotoxicity studies at low and medium dose levels in the rat. Arch Toxicol Suppl. 1984; 7:455–458.26. Chernikov VG, Terekhov SM, Krokhina TB, Shishkin SS, Smirnova TD, Kalashnikova EA, Adnoral NV, Rebrov LB, Denisov-Nikol'skii YI, Bykov VA. Comparison of cytotoxicity of aminoglycoside antibiotics using a panel cellular biotest system. Bull Exp Biol Med. 2003; 135:103–105.27. Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009; 52:2836–2845.28. Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, Leonard EM, Almstead NG, Ju W, Peltz SW, et al. Safety, tolerability, and pharmacokinetics of PTC124, a nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol. 2007; 47:430–444.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Heterozygous Mutations of The Gene for Kir 1.1 (ROMK) in Antenatal Bartter Syndrome Presenting with Transient Hyperkalemia, Evolving to a Benign Course
- A case of Bartter's syndrome
- A case of Bartter's syndrome
- A Case of Adult-Onset Bartter's Syndrome Associated with Nephrocalcinosis
- A Case of Bartter's Syndrome
