J Korean Med Sci.
2005 Dec;20(6):1076-1078. 10.3346/jkms.2005.20.6.1076.
Two Novel Mutations in the Aquaporin 2 Gene in a Girl with Congenital Nephrogenic Diabetes Insipidus
- Affiliations
-
- 1Department of Pediatrics, Seoul National University Children's Hospital, Seoul, Korea. cheonghi@plaza.snu.ac.kr
- 2Department of Pediatrics, Ewha Woman's University Dongdaemun Hospital, Seoul, Korea.
- 3Nephrology, Department of Internal Medicine, Yanbian University College of Medicine Yanbian Hospital, Yanji City, Jilin Prov., P.R. China.
- KMID: 2157767
- DOI: http://doi.org/10.3346/jkms.2005.20.6.1076
Abstract
- Congenital nephrogenic diabetes insipidus (CNDI) is a rare inherited disorder characterized by insensitivity of the kidney to the antidiuretic effect of vasopressin. There are three inheritance patterns of CNDI: the X-linked recessive form associated with vasopressin V2 receptor gene mutations, and the autosomal recessive and dominant forms associated with aquaporin-2 gene (AQP2) mutations. The evaluation for polyuria and polydipsia in a one-month-old Korean girl revealed no response to vasopressin and confirmed the diagnosis of CNDI. Because the child was female without family history of CNDI, her disease was thought to be an autosomal recessive form. We analyzed the AQP2 gene and detected a compound heterozygous missense point mutation: (70)Ala (GCC) to Asp (GAC) in exon 1 inherited from her father and (187)Arg (CGC) to His (CAC) in exon 3 inherited from her mother. The first mutation is located within the first NPA motif of the AQP2 molecule and the second one right after the second NPA motif. This is the first report to characterize AQP2 mutations in Korean patients with autosomal recessive CNDI, and expands the spectrum of AQP2 mutations by reporting two novel mutation, (70)Ala (GCC) to Asp (GAC) and (187)Arg (CGC) to His (CAC).
MeSH Terms
Figure
-
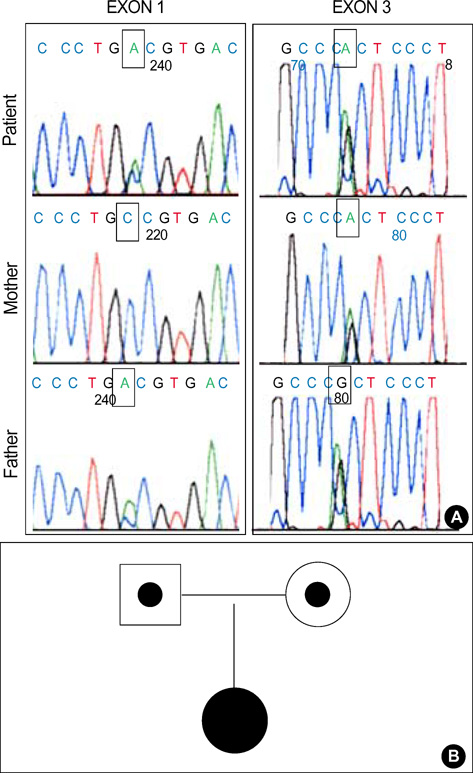
Fig. 1 (A) Direct sequence analysis at exon 1 and exon 3 of the aquaporin-2 genes originating from the patient, her mother and her father. The small squares indicate the sites of point mutation. (B) The pedigree showing autosomal recessive mode of inheritance.
Reference
-
1. Knoers NVAM, Deen PM. Molecular and cellular defects in nephrogenic diabetes insipidus. Pediatr Nephrol. 2001. 16:1146–1152.
Article2. Nomura Y, Onigata K, Nagashima T, Yutani S, Mochizuki H, Nagashima K, Morikawa A. Detection of skewed X-inactivation in two female carriers of vasopressin type 2 receptor gene mutation. J Clin Endocrinol Metab. 1997. 82:3434–3437.
Article3. van Lieburg AF, Knoers NVAM, Mallmann R, Proesmans W, van den Heuvel LP, Monnens LA. Normal fibrinolytic responses to 1-desamino-8-D-arginine vasopressin in patients with nephrogenic diabetes insipidus caused by mutations in the aquaporin-2 gene. Nephron. 1996. 72:544–546.4. Morello JP, Bichet DG. Nephrogenic diabetes insipidus. Annu Rev Physiol. 2001. 63:607–630.
Article5. Marr N, Bichet DG, Hoefs S, Savelkoul PJ, Konings IB, De Mattia F, Graat MP, Arthus MF, Lonergan M, Fujiwara TM, Knoers NV, Landau D, Balfe WJ, Oksche A, Rosenthal W, Muller D, Van Os CH, Deen PM. Cell-biologic and functional analyses of five new Aquaporin-2 missense mutations that cause recessive nephrogenic diabetes insipidus. J Am Soc Nephrol. 2002. 13:2267–2277.
Article6. Lin SH, Bichet DG, Sasaki S, Kuwahara M, Arthus MF, Lonergan M, Lin YF. Two novel aquaporin-2 mutations responsible for congenital nephrogenic diabetes insipidus in Chinese families. J Clin Endocrinol Metab. 2002. 87:2694–2700.
Article7. Marr N, Bichet DG, Lonergan M, Arthus MF, Jeck N, Seyberth HW, Rosenthal W, van Os CH, Oksche A, Deen PM. Heteroligomerization of an aquaporin-2 mutant with wild-type aquaporin-2 and their misrouting to late endosomes/lysosomes explains dominant nephrogenic diabetes insipidus. Hum Mol Genet. 2002. 11:779–789.
Article8. Kuwahara M, Iwai K, Ooeda T, Igarashi T, Ogawa E, Katsushima Y, Shinbo I, Uchida S, Terada Y, Arthus MF, Lonergan M, Fujiwara TM, Bichet DG, Marumo F, Sasaki S. Three families with autosomal dominant nephrogenic diabetes insipidus caused by aquaporin-2 mutations in the C-terminus. Am J Hum Genet. 2001. 69:738–748.
Article9. Verkman AS, Mitra AK. Structure and function of aquaporin water channels. Am J Physiol Renal Physiol. 2000. 278:F13–F28.
Article10. Deen PM, Verdijk MA, Knoers NV, Wieringa B, Monnens LA, van Os CH, van Oost BA. Requirement of human renal water channel aquaporin-2 for vasopressin-dependent concentration of urine. Science. 1994. 264:92–95.
Article11. Morello JP, Salahpour A, Laperriere A, Bernier V, Arthus MF, Lonergan M, Petaja-Repo U, Angers S, Morin D, Bichet DG, Bouvier M. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000. 105:887–895.
Article12. Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998. 101:2257–2267.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A case of primary aldosteronism combined with acquired nephrogenic diabetes insipidus
- A case of nephrogenic diabetes insipidus associated with urinary incontinence
- Congenital Nephrogenic Diabetes Insipidus Developing in the Mother and the Son
- Congenital Nephrogenic Diabetes Insipidus with Bilateal Hydronephrosis: Indomethacin in Treatment of Nephrogenic Diabetes Insipidus
- New insights into the transcriptional regulation of aquaporin-2 and the treatment of X-linked hereditary nephrogenic diabetes insipidus
