Effects of PPAR-alpha and-gamma Agonists on Fatty Acid Metabolism of Muscle Cells in Hyperlipidemic and Hyperglycemic Conditions
- Affiliations
-
- 1Yonsei University Brain Korea 21 Project for Medical Science, Korea.
- 2Division of Endocrinology and Metabolism, Department of Internal Medicine, Yonsei University College of Medicine, Korea.
- 3Department of Internal Medicine, Inha University College of Medicine, Korea.
- 4Department of Internal Medicine, College of Medicine, Pochon CHA University, Korea.
- 5Department of Endocrinology and Metabolism, Ajou Univerisity School of Medicine, Korea.
- 6Division of clinical research, Affiliated Research Institute, Seoul Medical Center, Korea.
- KMID: 2008262
- DOI: http://doi.org/10.4093/jkda.2006.30.5.324
Abstract
-
BACKGROUND: Studies for the regulation of fatty acid metabolism are deficient relatively in skeletal muscle and heart. The investigations in pathological conditions for malonyl-CoA decarboxylase (MCD) and for the relation of MCD and PPAR-alpha.-gamma agonists are insufficient in particular.
METHODS
In the current study, fully differentiated H9c2 muscle cells were exposed to pathological conditions such as hyperlipidemic (0.1 mM Palmitate) and hyperglycemic (16.5 mM Glucose) condition with 5 uM PPAR-gamma agonist (rosiglitazone) and 10 uM PPAR-alpha agonist (WY14,643) and then experiments such as MCD activity assay, MCD real-time RT-PCR, MCD reporter gene assay, MCD Western blotting, PPAR-alpha Western blotting, and palmitate oxidation test were carried out.
RESULTS
Only PPAR-alpha agonist increased MCD activity. In the result of real-time RT-PCR, both PPAR-alpha and PPAR-gamma agonists elevated MCD mRNA expression in hyperlipidemic condition. MCD protein expression was decreased in hyperlipidemic condition, however, increased in rosiglitazone, or WY14,643 treated conditions. Rosiglitazone, and WY14,643 treated groups showed incresed MCD protein expression in hyperglycemic condition. Hyperlipidemic control group and PPAR-alpha.-gamma agonists treated groups presented about 3.8 times more increased palmitate oxidation level than normolipidemic control group in hyperlipidemic condition. PPAR-alpha agonist treated group showed 49% more increased palmitate oxidation rate than hyperlipidemic control group in primary cultured rat skeletal muscle cells. The amount of palmitate oxidation from differentiated H9c2 muscle cells that had overexpressed PPAR-alpha structural genes was more increased than control group.
CONCLUSION
This study suggests that PPAR-alpha agonist ameliorates the defects induced by hyperlipidemic condition through the regulation of MCD. In summary, a closely reciprocal relation among PPAR-alpha agonist, MCD, and fatty acid oxidation existed distinctly in hyperlipidemic condition, but not in hyperglycemic condition.
Keyword
MeSH Terms
Figure
-
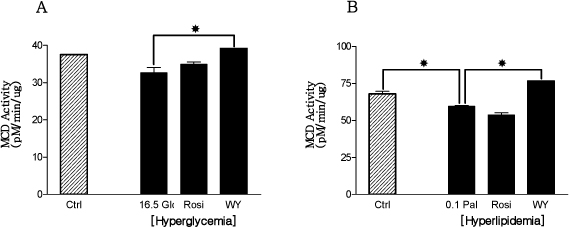
Fig. 1 The alteration of MCD activities in hyperglycemic (A) and hyperlipidemic (B) conditions. Fully differentiated myotubes were treated with 5 uM rosiglitazone and 10 uM WY14,643 in hyperglycemic condition and in hyperlipidemic condition. Only WY-14,643 increased MCD enzyme activity in hyperglycemic and hyperlipidemic conditions. *P < 0.05

Fig. 2 The alteration of MCD mRNA expressions in hyperglycemic (A) and hyperlipidemic (B) conditions. The elevation of MCD mRNA expression by PPAR agonists was observed in hyperlipidemic condition, however, not in hyperglycemic condition. *P < 0.05
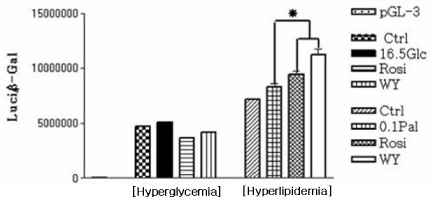
Fig. 3 The result of MCD promoter gene assay in hyperglycemic and hyperlipidemic conditions. The expression rate of MCD promoter gene was increased by PPAR agonists in hyperlipidemic condition, however, not in hyperglycemic condition. *P < 0.05
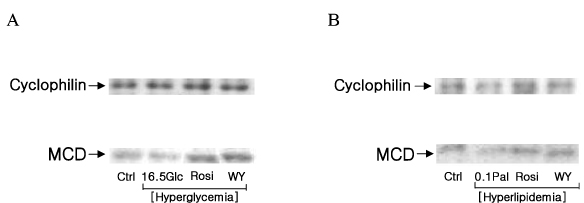
Fig. 4 The alteration of MCD protein expressions in hyperglycemic (A) and hyperlipidemic (B) conditions. MCD protein expression that was decreased in hyperlipidemic condition was increased by treatments of PPAR agonists. In hyperglycemic condition PPAR agonists increased MCD protein expression. *P < 0.05

Fig. 5 The effects of PPAR agonists on palmitate oxidations from H9c2 cells (A, B) in hyperglycemic (A) and hyperlipidemic (B) conditions, and from HepG2 cells (C) in hyperlipidemic condition. Palmitate oxidation was increased in only hyperlipidemic condition. The increase of palmitate oxidation from H9c2 cells occurred only in hyperlipidemic condition, but palmitate oxidation rates among hyperlipidemic control group and PPAR agonists treated groups were not different. HepG2 cells in hyperlipidemic condition was like to the palmitate oxidation aspect of H9c2 cells in same condition. *P < 0.05
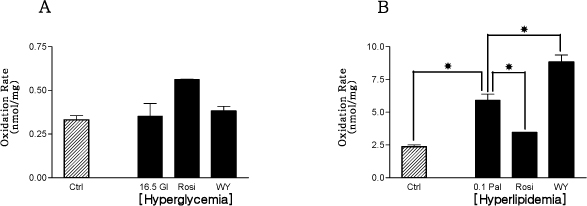
Fig. 6 The effects of PPAR agonists on palmitate oxidations from primary cultured rat skeletal muscle cell in hyperglycemic (A) and hyperlipidemic (B) conditions. There was no differences among experimental groups in hyperglycemic condition. But in hyperlipidemic condition the palmitate oxidation rates of hyperlipidemic control group and PPAR-α agonist treated group were more increased than normolipidemic control group and hyperlipidemic control group respectively, however, the palmitate oxidation rate of PPAR-γ agonist treated group was decreased in comparison with hyperlipidemic control group. *P < 0.05
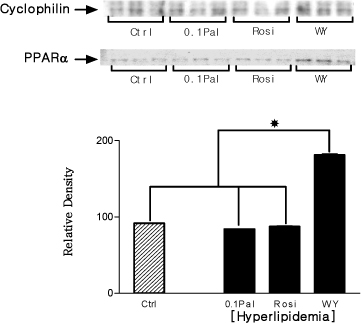
Fig. 7 The alteration of PPAR-α protein expression in hyperlipidemic condition In hyperlipidemic condition, only protein expression of WY14,643 treated group was increased. *P < 0.05.
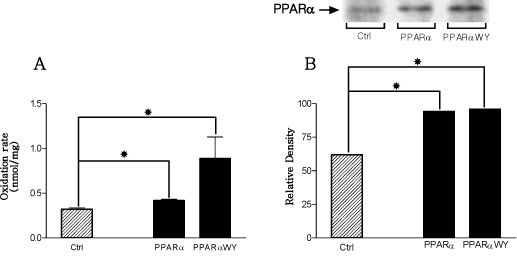
Fig. 8 The variations of palmitate oxidation (A) and PPAR-α protein expression (B) rates after PPAR-α structural gene overexpression. PPAR-α overexpression group showed increased palmitate oxidation rate and PPAR-α protein expression as comparison with control group. *P < 0.05.
Reference
-
1. Chien D, Dean D, Saha AK, Flatt JP, Ruderman NB. Malonyl-CoA content and fatty acid oxidation in rat muscle and liver in vivo. Am J Physiol Endocrinol Metab. 2000. 279:E259–E265.2. Dyck JRB, Berthiaume LG, Thomas PD, Kantor PK, Amy J, Barr AJ, Barr R. Characterization of rat liver malonyl-CoA decarboxylase and the study of its role in regulating fatty acid metabolism. Biochem J. 2000. 350:599–608.3. Alam N, Saggerson ED. Malonyl-CoA and the regulation of fatty acid oxidation in soleus muscle. Biochem J. 1998. 334:233–241.4. Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol. 1999. 276:E1–E18.5. Saha AK, Schwarsin AJ, Roduit R, Kaushik V, Tornheim K, Ruderman NB. Activation of malonyl-CoA decarboxylase in rat skeletal muscle by contraction and the AMP-activated protein kinase activator 5-Aminoimidazole-4-carboxamide-1-D-ribofuranoside. J Biol Chem. 2000. 275:24279–24283.6. Awan MM, Saggerson ED. Malonyl-CoA metabolism in cardiac myocytes and its relevance to the control of fatty acid oxidation. Biochem J. 1993. 295:61–66.7. Kudo N, Barr AJ, Barr RL, Desai S, Lopaschuk GD. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995. 270:17513–17520.8. Sakamoto J, Barr RL, Kavanagh KM, Lopaschuk GD. Conribution of malonyl-CoA decarboxylase to the high fatty acid oxidation rates seen in the diabetic heart. Am J Physiol Heart Circ Physiol. 2000. 278:H1196–H1204.9. Park HJ, Kaushik VK, Constant S, Prentki M, Przybytkowski E, Ruderman NB, Saha AK. Coordinate regulation of malonyl-CoA decarboxylase, sn-glycerol-3-phosphate acyltransferase, and acetyl-CoA carboxylase by AMP-activated protein kinase in rat tissues in response to exercise. J Biol Chem. 2002. 277:32571–32577.10. Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 2000. 37:1595–1607.11. Ruderman NB, Saha AK, Vavvas D, Witters LA. Malonyl-CoA, fuel sensing, and insulin resistance. Am J Physiol Endocrinol Metab. 1999. 276:E1–E18.12. Shimabukuro M, Zhou Y, Levi M, Unger R. Fatty acid-induced b-cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 1998. 95(5):2498–2502.13. Unger R, Zhou Y, Orci L. Regulation of fatty acid homeostasis in cells: novel role of leptin. Proc Natl Acad Sci USA. 1999. 96:2327–2332.14. Zhou Y, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger R. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA. 2000. 97:1784–1789.15. Young ME, Goodwin G, Ying J, Guthrie P, Christopher R, Wilson CR, Laws FA, Taegtmeyer H. Regulation of cardic and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am J Physiol Enocrinol Metab. 2001. 280:E471–E479.16. Latruffe N, Vamecq J. Peroxisome proliferators and peroxisome proliferator activated receptors (PPARs) as regulators of lipid metabolism. Biochimie. 1997. 79:81–94.17. Zinman B. PPARγ agonists in type2 diabetes: how far have we come in preventing the inevitable? A review of the metabolic effects of rosiglitazone. Diabetes Obes Metab. 2001. 3:S34–S43.18. Kramer D, Shapiro R, Adler A, Bush E, Rondinone CM. Insulin-sensitizing effect of rosiglitazone (BRL-49653) by regulation of glucose transporters in muscle and fat of Zucker rats. Metabolism. 2001. 50:1294–1300.19. Miyazaki Y, Mahankali A, Matsuda M, Glass L, Mahankali S, Ferrannini E, Cusi K, Mandarino LJ, DeFranzo RA. Improved glycemic control and enhanced insulin sensitivity in type 2 diabetic subjects treated with pioglitazone. Diabetes Care. 2001. 24:710–719.20. Spiegelman BM. PPAR-γ: Adipogenic regulator, thiazolidinedione receptor. Diabetes. 1998. 47:507–517.21. Chou CJ, Haluzik M, Gregory C, Dietz KR, Vinson C, Gavrilova O, Reitman ML. WY14643, a PPAR α Agonist, Improves Hepatic and Muscle and Steatosis and Reverses Insulin resistance in Lipoatrophic A-ZIP/F-1 Mice. J Biol Chem. 2002. 277:24484–24489.22. Lee GY, Cho JW, Lee HC, Kim YS. Genomic organization and characterization of the promoter of rat malonyl-CoA decarboxylase gene. Biochim Biophys Acta. 2002. 1577(1):133–138.23. Blau HM, Webster C. Isolation and characterization of human muscle cells. Proc Natl Acad Sci USA. 1981. 78:L5623–L5627.24. Sarabia V, Lam L, Burdett E, Leiter LA, Klip A. Glucose uptake in human and animall muscle cells in culture. Biochem Cell Biol. 1990. 68:536–542.25. Rognstad R. Estimation of peroxisomal and mitochondrial fatty acid oxidation in rat hepatocytes using tritiated substrates. Biochem J. 1991. 279:147–150.26. Young ME, Goodwin GW, Ying J, Guthrie P, Wilson CR, Laws FA, Taegtmeyer H. Regulation of cardiac and skeletal muscle malonyl-CoA decarboxylase by fatty acids. Am J Physiol Endocrinol Metab. 2001. 280:E471–E479.27. Lee GY, Kim NH, Zhao ZS, Cha BS, Kim YS. Peroxisomal-proliferator-activated receptor a activates transcription of the rat hepatic malonyl-CoA decarboxylase gene: a key regulation of malonyl-CoA level. Biochem J. 2004. 378:983–990.28. Campbell FM, Kozak R, Wagner A, Altarejos JY, Dyck JRB, Belke DD, Severson DL, Kelly DP, Lopaschuk GD. A Role for Peroxisome Proliferator-activated Receptorα (PPARα) in the Control of Cardiac Malonyl-CoA Levels. J Biol Chem. 2002. 277:4098–4103.29. Muoio DM, Way JM, Tanner CJ, Winegar DA, Kliewer SA, Houmard JA, Kraus WE, Dohm GL. Peroxisome Proliferator-Activated Receptor-α Regulates Fatty Acid Utilization in Primary Human Skeletal Muscle Cells. Diabetes. 2002. 51:901–909.30. Braissant O, Foufelle F, Scotto C, Dauca M, Wahle W. Differential expression of peroxisome proliferatoractivated receptor (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996. 137(1):354–366.31. Kim H, Haluzik M, Asghar Z, Yau D, Joseph JW, Fernandez AM, Reitman ML, Yakar S, Stannard B, Heron-Milhavet L, Wheeler MB, LeRoith D. Peroxisome Proliferator-Activated Receptor-α Agonisr Treatment in a Transgenic Model of Type 2 Diabetes Reverses the Lipotoxic State and Improves Glucose Homeostasis. Diabetes. 2003. 52:1770–1778.32. Koh EH, Kim MS, Park JY, Kim HS, Youn JY, Park HS, Youn JH, Lee KU. Peroxisome Proliferator-Activated Receptor (PPAR)-α Activation Prevents Diabetes in OLETF Rats: Comparison With PPARActivation. Diabetes. 2003. 52:2331–2337.33. Muoio DM, MacLean PS, Lang DB, Li S, Houmard JA, Way JM, Winegar DA, Corton JC, Dohm GL, Kraus WE. Fatty acid homeostasis and induction of lipid regulatory genes in skeletal muscles of peroxisome proliferator-activated receptor (PPAR) α knock-out mice. Evidence for compensatory regulation by PPAR delta. J Biol Chem. 2002. 277:26089–26097.34. Wolf G. The function of the nuclear receptor peroxisome proliferator-activated receptor delta in energy homeostasis. Nutr Rev. 2003. 61:387–390.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Safety and Efficacy of Peroxisome Proliferator-Activated Receptor-alpha Agonist for Treating Cardiovascular Disease
- Refocusing Peroxisome Proliferator Activated Receptor-alpha: A New Insight for Therapeutic Roles in Diabetes
- The Effects of Dietary Interventions on mRNA Expression of Peroxisome Proliferator Activated Receptor Isoforms (PPAR Isoforms)in Rat Skeletal Muscle
- Activation of PPARgamma induces profound multilocularization of adipocytes in adult mouse white adipose tissues
- Peroxisome Proliferator Activated Receptor-delta (PPAR-delta)
