Ann Lab Med.
2014 Sep;34(5):395-398. 10.3343/alm.2014.34.5.395.
Large Deletion in KCNQ1 Identified in a Family with Jervell and Lange-Nielsen Syndrome
- Affiliations
-
- 1Department of Laboratory Medicine, Seoul National University Hospital, Seoul, Korea. mwseong@snu.ac.kr
- 2Department of Pediatrics, Seoul National University Hospital, Seoul, Korea.
- 3Green Cross Laboratories, Seoul, Korea.
- 4Department of Laboratory Medicine, National Medical Center, Seoul, Korea.
- KMID: 1791957
- DOI: http://doi.org/10.3343/alm.2014.34.5.395
Abstract
- Long QT syndrome (LQTS) is a genetically heterogeneous disorder associated with sequence variations in more than 10 genes; in some cases, it is caused by large deletions or duplications among the main, known LQTS-associated genes. Here, we describe a 14-month-old Korean boy with congenital hearing loss and prolonged QT interval whose condition was clinically diagnosed as Jervell and Lange-Nielsen syndrome (JLNS), a recessive form of LQTS. Genetic analyses using sequence analysis and multiplex ligation-dependent probe amplification (MLPA) assay revealed a large deletion spanning exons 7-10 as well as a frameshift mutation (c.1893dup; p.Arg632Glnfs*20). To our knowledge, this is the first report of a large deletion in KCNQ1 identified in JLNS patients. This case indicates that a method such as MLPA, which can identify large deletions or duplications needs to be considered in addition to sequence analysis to diagnose JLNS.
Keyword
MeSH Terms
Figure
-

Fig. 1 Family pedigree. The older brother with a history of syncope and long QTc carries the same mutations as the proband (marked with arrow), i.e., large exon deletion and frameshift sequence mutation. Each mutation was inherited from one of the parents; the exon deletion from the father and the frameshift mutation from the mother.
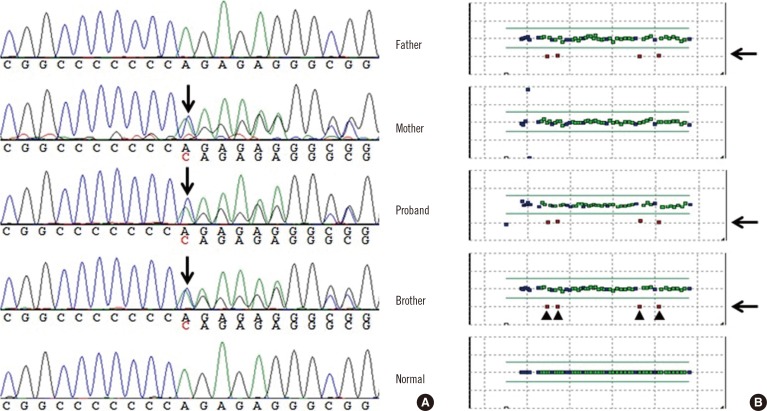
Fig. 2 (A) Sequencing analysis revealed a frameshift mutation (c.1893dup; p.Arg632Glnfs*20) in the proband, his mother, and his brother. (B) On the basis of the result of multiplex ligation-dependent probe amplification, 4 exons (exons 9, 10, 7, and 8 in this order) with half the copy number were identified, indicating a large deletion in the proband, his father, and his brother.
Reference
-
1. Kannankeril P, Fish F. Disorders of cardiac rhythm and conduction. In : Allen HD, Driscoll DJ, Shaddy RE, Feltes TF, editors. Moss and Adams' heart disease in infants, children, and adolescents including the fetus and youngadult. 7th ed. Philadelphia: Lippincott Williams & Wilkins;2008. p. 293–341.2. Baek JS, Bae EJ, Lee SY, Park SS, Kim SY, Jung KN, et al. Jervell and Lange-Nielsen syndrome: novel compound heterozygous mutations in the KCNQ1 in a Korean family. J Korean Med Sci. 2010; 25:1522–1525.3. Kanovsky J, Novotny T, Kadlecova J, Gaillyova R. A new homozygous mutation of the KCNQ1 gene associated with both Romano-Ward and incomplete Jervell Lange-Nielsen syndromes in two sisters. Heart Rhythm. 2010; 7:531–533.
Article4. Kaltenbach S, Capri Y, Rossignol S, Denjoy I, Soudée S, Aboura A, et al. Beckwith-Wiedemann syndrome and long QT syndrome due to familial-balanced translocation t(11;17)(p15.5;q21.3) involving the KCNQ1 gene. Clin Genet. 2013; 84:78–81.5. Tranebjaerg L, Bathen J, Tyson J, Bitner-Glindzicz M. Jervell and Lange-Nielsen syndrome: a Norwegian perspective. Am J Med Genet. 1999; 89:137–146.6. Goldenberg I, Moss AJ, Zareba W, McNitt S, Robinson JL, Qi M, et al. Clinical course and risk stratification of patients affected with the Jervell and Lange-Nielsen syndrome. J Cardiovasc Electrophysiol. 2006; 17:1161–1168.
Article7. Winbo A, Stattin EL, Diamant UB, Persson J, Jensen SM, Rydberg A. Prevalence, mutation spectrum, and cardiac phenotype of the Jervell and Lange-Nielsen syndrome in Sweden. Europace. 2012; 14:1799–1806. PMID: 22539601.
Article8. Tyson J, Tranebjaerg L, McEntagart M, Larsen LA, Christiansen M, Whiteford ML, et al. Mutational spectrum in the cardioauditory syndrome of Jervell and Lange-Nielsen. Hum Genet. 2000; 107:499–503.
Article9. Schwartz PJ, Spazzolini C, Crotti L, Bathen J, Amlie JP, Timothy K, et al. The Jervell and Lange-Nielsen syndrome: natural history, molecular basis, and clinical outcome. Circulation. 2006; 113:783–790.10. Neyroud N, Richard P, Vignier N, Donger C, Denjoy I, Demay L, et al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ Res. 1999; 84:290–297.11. Zehelein J, Kathoefer S, Khalil M, Alter M, Thomas D, Brockmeier K, et al. Skipping of Exon 1 in the KCNQ1 gene causes Jervell and Lange-Nielsen syndrome. J Biol Chem. 2006; 281:35397–35403.
Article12. Eddy CA, MacCormick JM, Chung SK, Crawford JR, Love DR, Rees MI, et al. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm. 2008; 5:1275–1281.
Article13. Wang Z, Li H, Moss AJ, Robinson J, Zareba W, Knilans T, et al. Compound heterozygous mutations in KvLQT1 cause Jervell and Lange-Nielsen syndrome. Mol Genet Metab. 2002; 75:308–316.
Article14. Ning L, Moss AJ, Zareba W, Robinson J, Rosero S, Ryan D, et al. Novel compound heterozygous mutations in the KCNQ1 gene associated with autosomal recessive long QT syndrome (Jervell and Lange-Nielsen syndrome). Ann Noninvasive Electrocardiol. 2003; 8:246–250.
Article15. Tester DJ, Benton AJ, Train L, Deal B, Baudhuin LM, Ackerman MJ. Prevalence and spectrum of large deletions or duplications in the major long QT syndrome-susceptibility genes and implications for long QT syndrome genetic testing. Am J Cardiol. 2010; 106:1124–1128.
Article16. Barc J, Briec F, Schmitt S, Kyndt F, Le Cunff M, Baron E, et al. Screening for copy number variation in genes associated with the long QT syndrome: clinical relevance. J Am Coll Cardiol. 2011; 57:40–47.17. Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000; 102:1178–1185.18. Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005; 2:507–517.
Article19. Richter S, Brugada P. Risk-stratifying Jervell and Lange-Nielsen syndrome from clinical data. J Cardiovasc Electrophysiol. 2006; 17:1169–1171.
Article20. Giudicessi JR, Ackerman MJ. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ Cardiovasc Genet. 2013; 6:193–200.21. Shinwari Z, Al-Hazzani A, Dzimiri N, Tulbah S, Mallawi Y, Al-Fayyadh M, et al. Identification of a novel KCNQ1 mutation in a large Saudi family with long QT syndrome: clinical consequences and preventive implications. Clin Genet. 2013; 83:370–374.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Jervell and Lange-Nielsen Syndrome: Novel Compound Heterozygous Mutations in the KCNQ1 in a Korean Family
- A Case of Congenital Long QT Syndrome Associated with Deafness and Syncope
- A Case of Congenital Long QT Syndrome with Reccurent Syncope
- Cornelia de Lange Syndrome
- Anesthetic Management of a Patient with Congenital Long QT Syndrome: A case report
