Clinical and Pathological Characteristics of Four Korean Patients with Limb-Girdle Muscular Dystrophy type 2B
- Affiliations
-
- 1Department of Neurology, Yongdong Severance Hospital, Seoul, Korea. ycchoi@yumc.yonsei.ac.kr
- 2Department of Rehabilitation Medicine, Yongdong Severance Hospital, Seoul, Korea.
- 3Department of Medicine, Euijong-bu Collectional Institution, Euijong-bu, Korea.
- 4Department of Pathology, Severance Hospital, Brain Korea 21 Project for Medicine, Yonsei University College of Medicine, Seoul, Korea.
- KMID: 1786826
- DOI: http://doi.org/10.3346/jkms.2004.19.3.447
Abstract
- Limb-girdle muscular dystrophy type 2B (LGMD2B), a subtype of autosomal recessive limb-girdle muscular dystrophy (ARLGMD), is characterized by a relatively late onset and slow progressive course. LGMD2B is known to be caused by the loss of the dysferlin protein at sarcolemma in muscle fibers. In this study, the clinical and pathological characteristics of Korean LGMD2B patients were investigated. Seventeen patients with ARLGMD underwent muscle biopsy and the histochemical examination was performed. For the immunocytochemistry, a set of antibodies against dystrophin, alpha, beta, gamma, delta-sarcoglycans, dysferlin, caveolin-3, and beta-dystroglycan was used. Four patients (24%) showed selective loss of immunoreactivity against dysferlin at the sarcolemma on the muscle specimens. Therefore, they were classified into the LGMD2B category. The age at the onset of disease ranged from 9 yr to 33 yr, and none of the patients was wheelchair bound at the neurological examination. The serum creatine kinase (CK) was high in all the patients (4010-5310 IU/L). The pathologic examination showed mild to moderate dystrophic features. These are the first Korean LGMD2B cases with a dysferlin deficiency confirmed by immunocytochemistry. The clinical, pathological, and immunocytochemical findings of the patients with LGMD2B in this study were in accordance with those of other previous reports.
Keyword
MeSH Terms
Figure
-
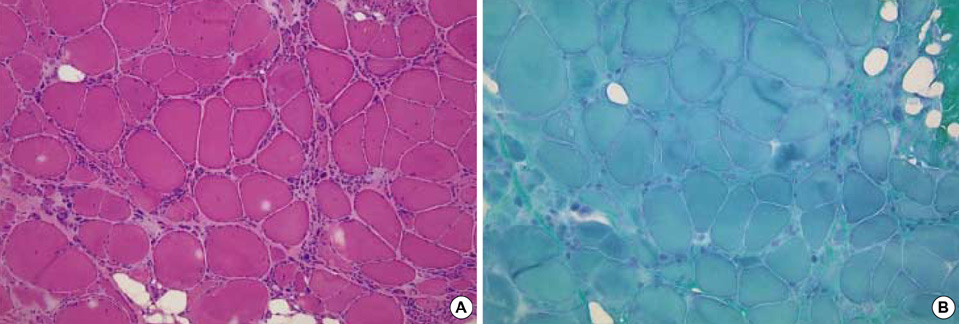
Fig. 1 Muscle specimens of the quadriceps femoris muscle (from patient No. 1) stained by hematoxylin-eosin (A) and modified Gomori Trichrome (B). There were great variation in the size of fibers, fiber splitting with internal nuclei, and clumps of small fibers (×200).
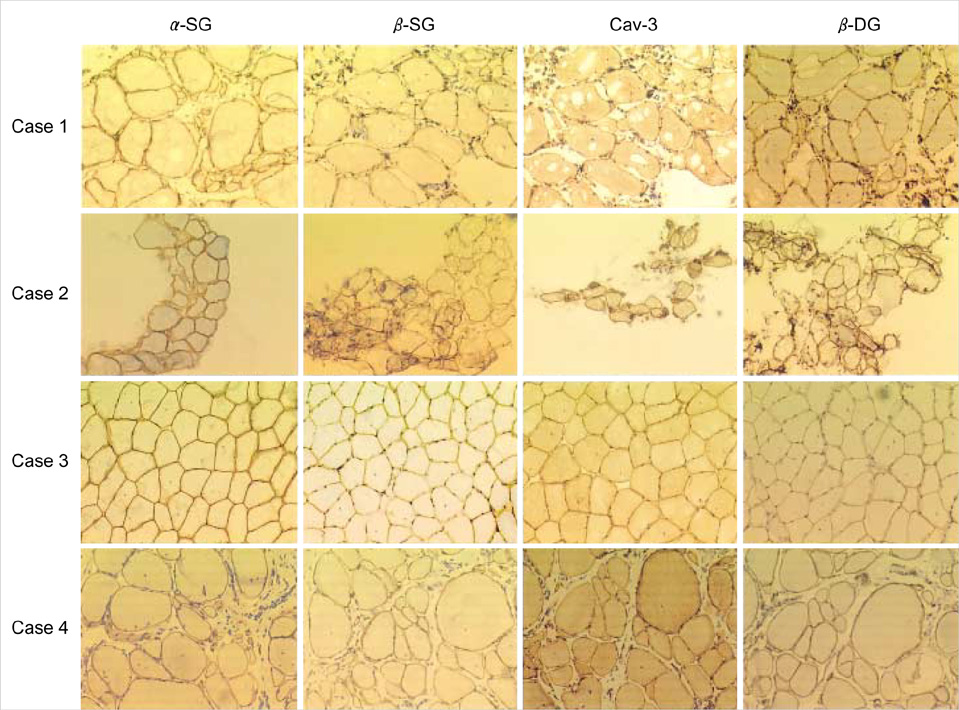
Fig. 2 Immunostaining against α-sarcoglycan (α-SG), β-sarcoglycan (β-SG), caveolin-3 (Cav-3), and β-dystroglycan (β-DG) on the muscle specimen in the four LGMD2B patients by the diaminobenzidine (DAB)-peroxidase method. All four LGMD2B patients showed a normal immunoreactivity against α-sarcoglycan, β-sarcoglycan, caveolin-3, and β-dystroglycan (×200).
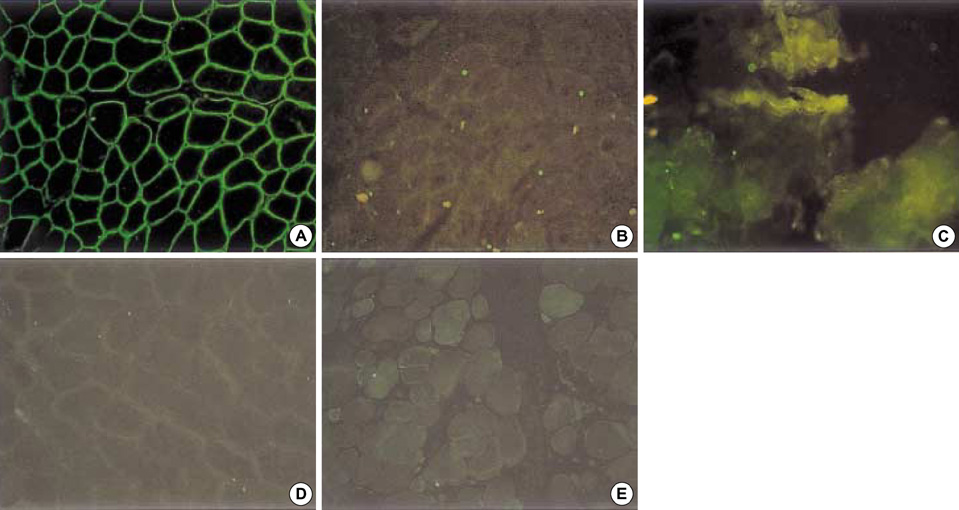
Fig. 3 Immunostaining against dysferlin on the muscle specimens from the four LGMD2B patients by a fluorescent isothiocyanate (FITC) technique. Normal control (A) showed a positive immunoreactivity against dysferlin at the sarcolemma. However, all four LGMD2B patients (B-E) showed a complete absence of a reaction against dysferlin at the sarcolemma on the muscle specimen (×200).
Reference
-
1. Zatz M, de Paula F, Starling A, Vainzof M. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord. 2003. 13:532–544.
Article2. Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH Jr. Dysferlin, a novel skeletal muscle genes, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998. 20:31–36.3. Britton S, Freeman T, Vafiadaki E, Keers S, Harrison R, Bushby K, Bashir R. The third human FER-1-like protein is highly similar to dysferlin. Genomics. 2000. 68:313–321.
Article4. Matsuda C, Aoki M, Hayashi YK, Ho MF, Arahata K, Brown RH Jr. Dysferlin is a surface membrane-associated protein that is absent in Miyoshi myopathy. Neurology. 1999. 53:1119–1122.
Article5. Anderson LV, Davison K, Moss JA, Young C, Cullen MJ, Walsh J, Johnson MA, Bashir R, Britton S, Keers S, Argov Z, Mahjneh I, Fougerousse F, Beckmann JS, Bushby KM. Dysferlin is a plasma membrane protein and is expressed early in human development. Hum Mol Genet. 1999. 8:855–861.
Article6. Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003. 423:168–172.
Article7. Weiler T, Greenberg CR, Nylen E, Halliday W, Morgan K, Eggertson D, Wrogemann K. Limb-girdle muscular dystrophy and Miyoshi myopathy in an aboriginal Canadian kindred map to LGMD2B and segregate with the same haplotype. Am J Hum Genet. 1996. 59:872–878.8. Illarioshkin SN, Ivanova-Smolenskaya IA, Tanaka H, Vereshchagin NV, Markova ED, Poleshchuk VV, Lozhnikova SM, Sukhorukov VS, Limborska SA, Slominsky PA, Bulayeva KB, Tsuji S. Clinical and molecular analysis of a large family with three distinct phenotypes of progressive muscular dystrophy. Brain. 1996. 119:1895–1909.
Article9. Illarioshkin SN, Ivanova-Smolenskaya IA, Greenberg CR, Nylen E, Sukhorukov VS, Poleshchuk VV, Markova ED, Wrogemann K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology. 2000. 55:1931–1933.
Article10. Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, Nylen E, Keers S, Vafiadaki E, Greenberg CR, Bushby CR, Wrogemann K. Identical mutation in patients with limb girdle muscular dystrophy type 2B or Miyoshi myopathy suggests a role for modifier gene(s). Hum Mol Genet. 1999. 8:871–877.
Article11. Linssen WH, Notermans NC, Van der Graaf Y, Wokke JH, Van Doorn PA, Howeler CJ, Busch HF, De Jager AE, De Visser M. Miyoshi-type distal muscular dystrophy. Clinical spectrum in 24 Dutch patients. Brain. 1997. 120:1989–1996.12. Bushby KM. Making sense of the limb-girdle muscular dystrophies. Brain. 1999. 122:1403–1420.
Article13. Choi YC, Park GT, Kim TS, Sunwoo IN, Steinert PM, Kim SY. Sporadic inclusion body myositis correlates with increased expression and cross-linking by transglutaminases 1 and 2. J Biol Chem. 2000. 275:8703–8710.
Article14. Bushby KM. The limb-girdle muscular dystrophies-multiple genes, multiple mechanisms. Hum Mol Genet. 1999. 8:1875–1882.15. Passos-Bueno MR, Vainzof M, Moreira ES, Zatz M. Seven autosomal recessive limb-girdle muscular dystrophies in the Brazilian population: from LGMD2A to LGMD2G. Am J Med Genet. 1999. 82:392–398.
Article16. Ueyama H, Kumamoto T, Horinouchi H, Fujimoto S, Aono H, Tsuda T. Clinical heterogeneity in dysferlinopathy. Intern Med. 2002. 41:532–536.
Article17. Argov Z, Sadeh M, Mazor K, Soffer D, Kahana E, Eisenberg I, Mitrani-Rosenbaum S, Richard I, Beckmann J, Keers S, Bashir R, Bushby K, Rosenmann H. Muscular dystrophy due to dysferlin deficiency in Libyan Jews. Clinical and genetic features. Brain. 2000. 123:1229–1237.18. Zatz M, Vainzof M, Passos-Bueno MR. Limb-girdle muscular dystrophy: one gene with different phenotypes, one phenotype with different genes. Curr Opin Neurol. 2000. 13:511–517.
Article19. Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH Jr. Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003. 278:50466–50473.
Article20. Tagawa K, Ogawa M, Kawabe K, Yamanaka G, Matsumura T, Goto K, Nonaka I, Nishino I, Hayashi YK. Protein and gene analyses of dysferlinopathy in a large group of Japanese muscular dystrophy patients. J Neurol Sci. 2003. 211:23–28.
Article21. Fanin M, Pegoraro E, Matsuda-Asada C, Brown RH Jr, Angelini C. Calpain-3 and dysferlin protein screening in patients with limb-girdle dystrophy and myopathy. Neurology. 2001. 56:660–665.
Article22. Vainzof M, Anderson LV, McNally EM, Davis DB, Faulkner G, Valle G, Moreira ES, Pavanello RC, Passos-Bueno MR, Zatz M. Dysferlin protein analysis in limb-girdle muscular dystrophies. J Mol Neurosci. 2001. 17:71–80.
Article23. Anderson LV, Harrison RM, Pogue R, Vafiadaki E, Pollitt C, Davison K, Moss JA, Keers S, Pyle A, Shaw PJ, Mahjneh I, Argov Z, Greenberg CR, Wrogemann K, Bertorini T, Goebe HH, Beckmann JS, Bashir R, Bushby KM. Secondary reduction in calpain 3 expression in patients with limb girdle muscular dystrophy type 2B and Miyoshi myopathy (primary dysferlinopathies). Neuromuscul Disord. 2000. 10:553–559.
Article24. Aoki M, Liu J, Richard I, Bashir R, Britton S, Keers SM, Oeltjen J, Brown HE, Marchand S, Bourg N, Beley C, McKenna-Yasek D, Arahata K, Bohlega S, Cupler E, Illa I, Majneh I, Barohn RJ, Urtizberea JA, Fardeau M, Amato A, Angelini C, Bushby K, Beckmann JS, Brown RH Jr. Genomic organization of the dysferlin gene and novel mutations in Miyoshi myopathy. Neurology. 2001. 57:271–278.
Article25. Piccolo F, Moore SA, Ford GC, Campbell KP. Intracellular accumulation and reduced sarcolemmal expression of dysferlin in limb-girdle muscular dystrophies. Ann Neurol. 2000. 48:902–912.
Article26. Matsuda C, Hayashi YK, Ogawa M, Aoki M, Murayama K, Nishino I, Nonaka I, Arahata K, Brown RH Jr. The sarcolemmal proteins dysferlin and caveolin-3 interact in skeletal muscle. Hum Mol Genet. 2001. 10:1761–1766.
Article
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Early-Onset LMNA-Associated Muscular Dystrophy with Later Involvement of Contracture
- Limb-Girdle Muscular Dystrophy
- Heterogeneous Characteristics of Korean Patients with Dysferlinopathy
- Mutations of CAPN3 in Korean Patients with Limb-Girdle Muscular Dystrophy
- Progressive Muscular Dystrophy (Report of 32 cases)
