J Korean Med Sci.
2012 Oct;27(10):1273-1277. 10.3346/jkms.2012.27.10.1273.
A Case of 9.7 Mb Terminal Xp Deletion Including OA1 Locus Associated with Contiguous Gene Syndrome
- Affiliations
-
- 1Greencross Reference Laboratory, Seoul, Korea.
- 2Department of Ophthalmology, Catholic University of Daegu School of Medicine, Daegu, Korea.
- 3Department of Pediatrics, Catholic University of Daegu School of Medicine, Daegu, Korea. kimjk@cu.ac.kr
- KMID: 1778839
- DOI: http://doi.org/10.3346/jkms.2012.27.10.1273
Abstract
- Terminal or interstitial deletions of Xp (Xp22.2-->Xpter) in males have been recognized as a cause of contiguous gene syndromes showing variable association of apparently unrelated clinical manifestations such as Leri-Weill dyschondrosteosis (SHOX), chondrodysplasia punctata (CDPX1), mental retardation (NLGN4), ichthyosis (STS), Kallmann syndrome (KAL1), and ocular albinism (GPR143). Here we present a case of a 13.5 yr old boy and sister with a same terminal deletion of Xp22.2 resulting in the absence of genes from the telomere of Xp to GPR143 of Xp22. The boy manifested the findings of all of the disorders mentioned above. We began a testosterone enanthate monthly replacement therapy. His sister, 11 yr old, manifested only Leri-Weill dyschondrosteosis, and had engaged in growth hormone therapy for 3 yr. To the best of our knowledge, this is the first report of a male with a 9.7 Mb terminal Xp deletion including the OA1 locus in Korea.
Keyword
MeSH Terms
-
Abnormalities, Multiple/*genetics
Adolescent
Child
Chromosome Deletion
*Chromosomes, Human, X
Eye Proteins/genetics
Female
Genetic Loci
Growth Hormone/therapeutic use
Humans
Male
Membrane Glycoproteins/genetics
Telomere/genetics
WAGR Syndrome/*diagnosis/genetics/therapy
Eye Proteins
Membrane Glycoproteins
Growth Hormone
Figure
-
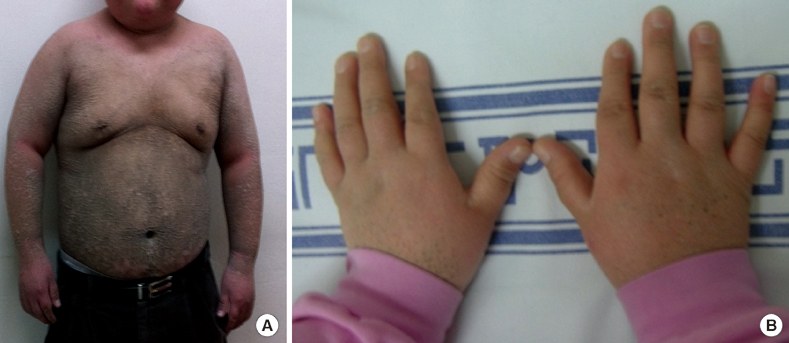
Fig. 1 Clinical features of the male patient. (A) Ichthyosis of trunk. (B) Shortness of terminal phlanges.

Fig. 2 Clinical features of his sister: rhizomeric and mesomelic shortening of forearms.
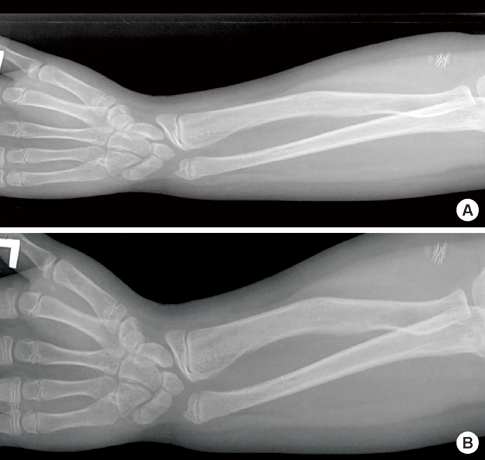
Fig. 3 Forearm radiographies of the male patient (A) and his sister (B), showing Madelung deformity.
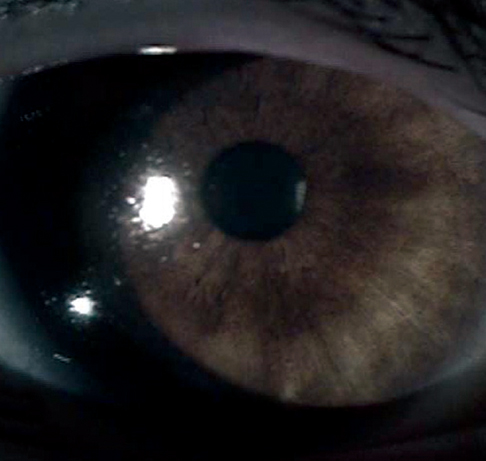
Fig. 4 Slit lamp photograph of defects in the pigmented iris epithelium.
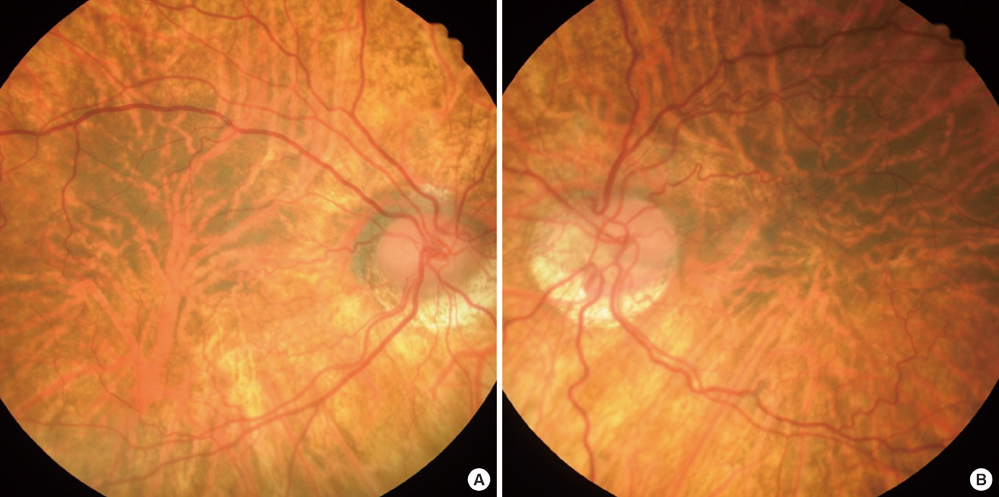
Fig. 5 Funduscopic photographs showing defective fundus pigmentation with macular hypoplasia in both eyes. (A) Right, (B) Left eye.

Fig. 6 Physical map of the region Xp22.2-pterminal: High resolution microarray analysis revealed a terminal 9.7 Mb deletion at Xp22.2 resulting absence of genes from the telomere of Xp to GPR143 of Xp22.
Reference
-
1. Ballabio A, Bardoni B, Carrozzo R, Andria G, Bick D, Campbell L, Hamel B, Ferguson-Smith MA, Gimelli G, Fraccaro M, et al. Contiguous gene syndromes due to deletions in the distal short arm of the human X chromosome. Proc Natl Acad Sci USA. 1989. 86:10001–10005.2. Ballabio A, Andria G. Deletions and translocations involving the distal short arm of the human X chromosome: review and hypotheses. Hum Mol Genet. 1992. 1:221–227.3. Melichar VO, Guth S, Hellebrand H, Meindl A, von der Hardt K, Kraus C, Trautmann U, Rascher W, Rauch A, Zenker M. A male infant with a 9.6 Mb terminal Xp deletion including the OA1 locus: Limit of viability of Xp deletions in males. Am J Med Genet A. 2007. 143:135–141.4. Meindl A, Hosenfeld D, Brückl W, Schuffenhauer S, Jenderny J, Bacskulin A, Oppermann HC, Swensson O, Bouloux P, Meitinger T. Analysis of a terminal Xp22.3 deletion in a p atient with six monogenic disorders: implications for the mapping of X linked ocular albinism. J Med Genet. 1993. 30:838–842.5. Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. The DNA sequence of the human X chromosome. Nature. 2005. 434:325–337.6. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005. 434:400–404.7. Rao E, Weiss B, Fukami M, Rump A, Niesler B, Mertz A, Muroya K, Binder G, Kirsch S, Winkelmann M, et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat Genet. 1997. 16:54–63.8. Belin V, Cusin V, Viot G, Girlich D, Toutain A, Moncla A, Vekemans M, Le Merrer M, Munnich A, Cormier-Daire V. SHOX mutations in dyschondrosteosis (Leri-Weill syndrome). Nat Genet. 1998. 19:67–69.9. Shears DJ, Vassal HJ, Goodman FR, Palmer RW, Reardon W, Superti-Furga A, Scambler PJ, Winter RM. Mutation and deletion of the pseudoautosomal gene SHOX cause Leri-Weill dyschondrosteosis. Nat Genet. 1998. 19:70–73.10. Binder G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm Res Paediatr. 2011. 75:81–89.11. Franco B, Meroni G, Parenti G, Levilliers J, Bernard L, Gebbia M, Cox L, Maroteaux P, Sheffield L, Rappold GA, et al. A cluster of sulfatase genes on Xp22.3: mutations in chondrodysplasia punctata (CDPX) and implications for warfarin embryopathy. Cell. 1995. 81:15–25.12. Lonardo F, Parenti G, Luquetti DV, Annunziata I, Della Monica M, Perone L, De Gregori M, Zuffardi O, Brunetti-Pierri N, Andria G, et al. Contiguous gene syndrome due to an interstitial deletion in Xp22.3 in a boy with ichthyosis, chondrodysplasia punctata, mental retardation and ADHD. Eur J Med Genet. 2007. 50:301–308.13. Jamain S, Quach H, Betancur C, Råstam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, Bourgeron T. Paris Autism Research International Sibpair Study. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003. 34:27–29.14. Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004. 74:552–557.15. Bonifas JM, Morley BJ, Oakey RE, Kan YW, Epstein EH Jr. Cloning of a cDNA for steroid sulfatase: frequent occurrence of gene deletions in patients with recessive X chromosome-linked ichthyosis. Proc Natl Acad Sci USA. 1987. 84:9248–9251.16. Roux ND, Young J, Misrahi M, Genet R, Chanson P, Schaison G, Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997. 337:1597–1602.17. Charles SJ, Green JS, Grant JW, Yates JR, Moore AT. Clinical features of affected males with X linked ocular albinism. Br J Ophthalmol. 1993. 77:222–227.18. Tobias ES, Bryce G, Farmer G, Barton J, Colgan J, Morrison N, Cooke A, Tolmie JL. Absence of learning difficulties in a hyperactive boy with a terminal Xp deletion encompassing the MRX49 locus. J Med Genet. 2001. 38:466–470.19. Boycott KM, Parslow MI, Ross JL, Miller IP, Bech-Hansen NT, MacLeod PM. A familial contiguous gene deletion syndrome at Xp22.3 characterized by severe learning disabilities and ADHD. Am J Med Genet A. 2003. 122A:139–147.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A female patient with Xp21 gene deletion syndrome
- Two Cases of Xp21 Contiguous Gene Deletion Syndrome
- Terminal Deletion of the Chromosome 4q with Hemivertebra: Case Report
- A Case of Xp21 Contiguous Gene Deletion Syndrome with Hyperglycerolemia, Congenital Adrenal Hypoplasia and Duchenne Muscular Dystrophy
- X-linked Hypophosphatemic Rickets, del(2)(q37.1;q37.3) Deletion Syndrome and Mosaic Turner Syndrome, mos 45,X/46,X, del(2)(q37.1;q37.3) in a 3-year-old Female
