Endocrinol Metab.
2012 Jun;27(2):151-154. 10.3803/EnM.2012.27.2.151.
A Case of CATCH22 Syndrome with Normal Parathyroid Function
- Affiliations
-
- 1Department of Endocrinology and Metabolism, Ajou University School of Medicine, Suwon, Korea. yschung@ajou.ac.kr
- 2Department of Medical Genetics, Ajou University School of Medicine, Suwon, Korea.
- KMID: 1497656
- DOI: http://doi.org/10.3803/EnM.2012.27.2.151
Abstract
- CATCH 22 is a medical acronym for cardiac defects, abnormal faces, thymic hypoplasia, cleft palate, and hypocalcemia, and a variable deletion on chromosome 22. It includes DiGeorge syndrome, conotruncal anomaly face syndrome, and velo-cardio-facial syndrome. It has a prevalence estimated at 1:3,000-1:6,000. Most deletions occur at de novo, but autosomal dominant inheritance is observed in 6-10% of cases. Hormonal disorders are common in patients with CATCH22 syndrome. While hypoparathyroidism was the predominant endocrine disturbance that has been documented in the DiGeorge syndrome, other hormonal defects, such as growth hormone deficiency, hypothyroidism, and hyperthyroidism have been occurred in patients with CATCH22 syndrome. The spectrum of parathyroid gland dysfunction in this syndrome ranges from severe neonatal hypocalcemia to normal parathyroid function. Most patients are usually diagnosed in young age, but a few patients with mild abnormality are presented later in life. We report a case of CATCH22 syndrome with normal parathyroid hormone and calcium level in an adult. The diagnosis of CATCH22 syndrome was confirmed by fluorescence in situ hybridization analysis.
Keyword
MeSH Terms
Figure
-
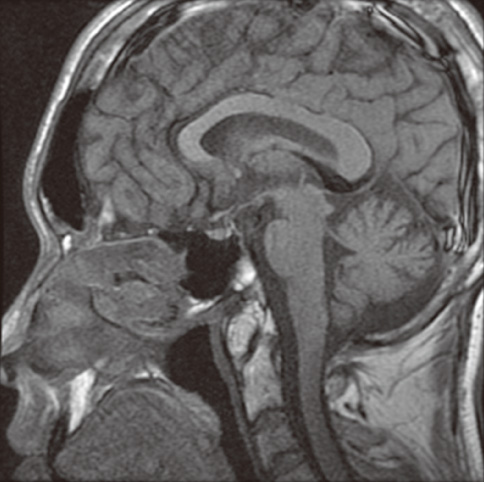
Fig. 1 Brain magnetic resonance imaging showed normal findings. There is no abnormal lesion in the cerebrum and cerebellum including sellar area.
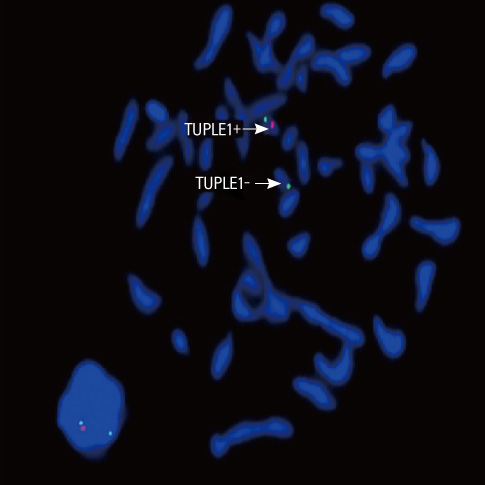
Fig. 2 Result of fluorescence in situ hybridization analysis during metaphase. Red spot is TUPLE1 gene probe (DiGeorge/velo-cardio-facial syndrome region of chromosome 22q11.2) and green spot is ARSA control region (22q13.3) probe. Red spot is visible only in one of chromosomes number 22.
Cited by 1 articles
-
A Case of CATCH22 Syndrome Diagnosed in Postmenopausal Woman
Seung Kyung Lee, Min Jeong Lee, Hyo Jin Lee, Bu Kyung Kim, Young Bae Sohn, Yoon-Sok Chung
J Bone Metab. 2013;20(1):57-60. doi: 10.11005/jbm.2013.20.1.57.
Reference
-
1. Wilson DI, Britton SB, McKeown C, Kelly D, Cross IE, Strobel S, Scambler PJ. Noonan's and DiGeorge syndromes with monosomy 22q11. Arch Dis Child. 1993. 68:187–189.2. Robin NH, Shprintzen RJ. Defining the clinical spectrum of deletion 22q11.2. J Pediatr. 2005. 147:90–96.3. Driscoll DA, Salvin J, Sellinger B, Budarf ML, McDonald-McGinn DM, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DiGeorge and velocardiofacial syndromes: implications for genetic counselling and prenatal diagnosis. J Med Genet. 1993. 30:813–817.4. Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007. 370:1443–1452.5. Jung SM, Bae JH, Kim DH, Na BG, Oh TG, Kim DW, Cho MC. A case of DiGeorge syndrome associated with complex cardiovascular anomalies. Korean J Med. 1997. 53:714–719.6. Kim DJ, Hyun YY, Choi HM, Lee JE, Kwon YJ, Pyo HJ, Choi EJ. DiGeorge syndrome diagnosed by hypocalcemic tetany. Korean J Med. 2006. 70:S299–S301.7. Lim JS, Hwang JS, Lee JA, Kim DH, Park KD, Cheon GJ, Shin CH, Yang SW. Bone mineral density according to age, bone age, and pubertal stages in Korean children and adolescents. J Clin Densitom. 2010. 13:68–76.8. Stevens CA, Carey JC, Shigeoka AO. DiGeorge anomaly and velocardiofacial syndrome. Pediatrics. 1990. 85:526–530.9. Balci S, Altugan FS, Alehan D, Aypar E, Baltaci V. A prenatally sonographically diagnosed conotruncal anomaly with mosaic type trisomy 21 and 22q11.2 microdeletion/DiGeorge syndrome. Genet Couns. 2009. 20:373–377.10. Choi JH, Shin YL, Kim GH, Seo EJ, Kim Y, Park IS, Yoo HW. Endocrine manifestations of chromosome 22q11.2 microdeletion syndrome. Horm Res. 2005. 63:294–299.11. Brauner R, Le Harivel de Gonneville A, Kindermans C, Le Bidois J, Prieur M, Lyonnet S, Souberbielle JC. Parathyroid function and growth in 22q11.2 deletion syndrome. J Pediatr. 2003. 142:504–508.12. Weinzimer SA, McDonald-McGinn DM, Driscoll DA, Emanuel BS, Zackai EH, Moshang T Jr. Growth hormone deficiency in patients with 22q11.2 deletion: expanding the phenotype. Pediatrics. 1998. 101:929–932.13. Chinen J, Rosenblatt HM, Smith EO, Shearer WT, Noroski LM. Long-term assessment of T-cell populations in DiGeorge syndrome. J Allergy Clin Immunol. 2003. 111:573–579.14. Karayiorgou M, Simon TJ, Gogos JA. 22q11.2 microdeletions: linking DNA structural variation to brain dysfunction and schizophrenia. Nat Rev Neurosci. 2010. 11:402–416.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- A Case of CATCH22 Syndrome with First Attack of Hypocalcemic Seizure at 13 Years of Age
- A Case of CATCH22 Syndrome Diagnosed in Postmenopausal Woman
- Tips for Preservation of Parathyroid Gland During Thyroid Surgery
- A Case of Hungry Bone Syndrome after Removal of a Parathyroid Adenoma
- Parathyroid Adenoma without Hyperparathyroidism Presenting as a Large Neck Mass
