A Practical Approach to Genetic Hypokalemia
- Affiliations
-
- 1Division of Nephrology, Department of Medicine, Tri-Service General Hospital, National Defense Medical Center, Taipei, Taiwan, Republic of China. l521116@ndmctsgh.edu.tw
- 2Department of Medicine, Providence St. Vincent Medical Center, Portland, Oregon, USA.
- KMID: 1463001
- DOI: http://doi.org/10.5049/EBP.2010.8.1.38
Abstract
- Mutations in genes encoding ion channels, transporters, exchangers, and pumps in human tissues have been increasingly reported to cause hypokalemia. Assessment of history and blood pressure as well as the K+ excretion rate and blood acid-base status can help differentiate between acquired and inherited causes of hypokalemia. Familial periodic paralysis, Andersen's syndrome, congenital chloride-losing diarrhea, and cystic fibrosis are genetic causes of hypokalemia with low urine K+ excretion. With respect to a high rate of K+ excretion associated with faster Na+ disorders (mineralocorticoid excess states), glucoricoid-remediable aldosteronism and congenital adrenal hyperplasia due to either 11beta-hydroxylase and 17alpha-hydroxylase deficiencies in the adrenal gland, and Liddle's syndrome and apparent mineralocorticoid excess in the kidney form the genetic causes. Among slow Cl- disorders (normal blood pressure, low extracellular fluid volume), Bartter's and Gitelman's syndrome are most common with hypochloremic metabolic alkalosis. Renal tubular acidosis caused by mutations in the basolateral Na+/HCO3 - cotransporter (NBC1) in the proximal tubules, apical H+-ATPase pump, and basolateral Cl-/HCO3 - exchanger (anion exchanger 1, AE1) in the distal tubules and carbonic anhydroase II in both are genetic causes with hyperchloremic metabolic acidosis. Further work on genetic causes of hypokalemia will not only provide a much better understanding of the underlying mechanisms, but also set the stage for development of novel therapies in the future.
Keyword
MeSH Terms
-
Acid-Base Equilibrium
Acidosis
Acidosis, Renal Tubular
Adrenal Glands
Adrenal Hyperplasia, Congenital
Aldosterone
Alkalosis
Blood Pressure
Carbon
Cystic Fibrosis
Diarrhea
Extracellular Fluid
Humans
Hyperaldosteronism
Hypokalemia
Hypotension
Ion Channels
Kidney
Mineralocorticoid Excess Syndrome, Apparent
Paralyses, Familial Periodic
Renin
Aldosterone
Carbon
Ion Channels
Mineralocorticoid Excess Syndrome, Apparent
Renin
Figure
-
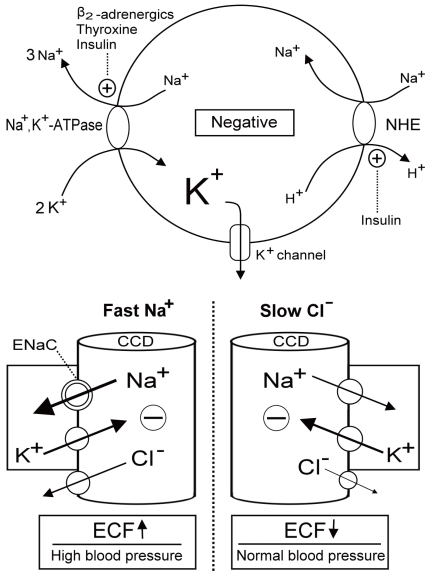
Fig. 1 Regulation of K+ redistribution in cells and K+ secretion in the cortical collecting duct (CCD). The circle depicts the cell membrane (upper panel). Na+,K+-ATPase, Na+/H+ exchanger (NHE), and K+ channels are three major elements controlling K+ shift. Na+,K+-ATPase is activated by β2-adrenergics, insulin and thyroid hormone. NHE, which causes the electroneutral entry of Na+ into cells and thus the net exit of positive voltage via the Na+,K+-ATPase, is also activated by insulin. K+ channels which permit K+ exit is responsible for generating the majority of the resting membrane potential and blocked by barium. The barrel shaped structures represent the terminal CCD (lower panel). The reabsorption of Na+ faster than Cl- (right) or Cl- slower than Na+ (left) in the CCD creates the lumen negative voltage that drives the net secretion of K+. Fast Na+ disorders cause extracellular fluid (ECF) volume expansion and high blood pressure, whereas, slow Cl- disorders lead to diminished ECF volume and low to normal blood pressure. ENaC, epithelial Na+ channels.
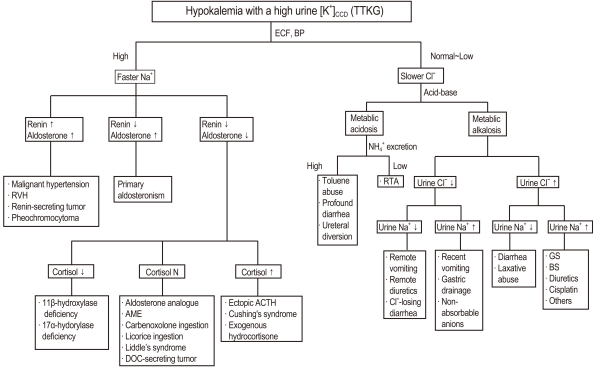
Fig. 2 Algorithm for the approach to hypokalemia with a high urine [K+]CCD (TTKG). CCD, cortical collecting duct; TTKG, transtubular K+ gradient; ECF, extracellular fluid; BP, blood pressure; RVH, right ventricular hypertrophy; AME, Apparent mineralocorticoid excess; DOC, 11-deoxycorticosterone; ACTH, adrenocorticotrophic hormone; RTA, renal tubular acidosis; GS, Gitelman's syndrome; BS, Bartter's syndrome
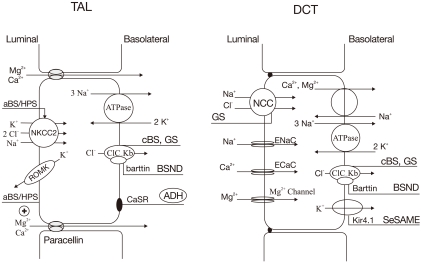
Fig. 3 Transport proteins in the thick ascending limb (TAL) of loop of Henle (LOH) (left panel) and distal convoluted tubule (DCT) (right panel) affected by gene mutations. Mutations that inactivate Na+/K+/2Cl- cotransporter (NKCC2) or renal outer medullary K+ channel (ROMK) lead to antenatal Bartter syndrome/hyperprostaglandin E syndrome (aBS/HPS) (BS type I and II) and mutations inactivating ClCKb cause classic Bartter syndrome (cBS) (BS type III). Mutations in barttin (chloride channel βsubunit) cause antenatal BS with sensorineural deafness (BSND) (BS type IV). Mutations that activate the calcium-sensing receptors (CaSR) occur in patients with autosomal dominant hypoparathyroidism (ADH) (BS type V). Mutations inactivating Na+/Cl- cotransporter (NCC) or basolateral Cl- channel (ClC-Kb) can cause Gitelman's syndrome (GS). Mutations in Kir4.1 channel cause seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME) syndrome. ADH, anti-diuretic hormone
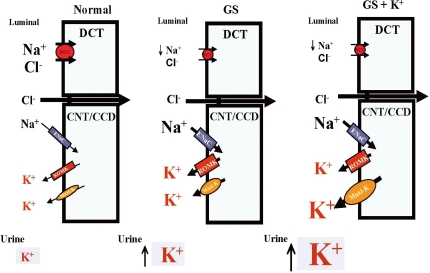
Fig. 4 Mechanisms for persistent hypokalemia in Gitelman's syndrome (GS). Enhanced epithelial Na+ channels (ENaC), renal outer medullary K+ channel (ROMK)1 and Maxi-K expression leads to hypokalemia (middle). High K+ supplement stimulates more accentuated maxi-K expression, accounting for persistent hypokalemia (left). DCT, distal convoluted tubule; CNT, cortical connecting tubules; CCD, cortical colleting duct.
Reference
-
1. Lin SH, Halperin ML. Hypokalemia: a practical approach to diagnosis and its genetic basis. Curr Med Chem. 2007; 14:1551–1565. PMID: 17584063.
Article2. Scheinman SJ, Guay-Woodford LM, Thakker RV, Warnock DG. Genetic disorders of renal electrolyte transport. N Engl J Med. 1999; 340:1177–1187. PMID: 10202170.
Article4. Sterns RH, Cox M, Feig PU, Singer I. Internal potassium balance and the control of the plasma potassium concentration. Medicine. 1981; 60:339–354. PMID: 6268928.
Article5. Clausen T. Clinical and therapeutic significance of the Na+, K+ pump. Clin Sci. 1998; 95:3–17. PMID: 9662481.6. Shieh CC, Coghlan M, Sullivan JP, Gopalakrishnan M. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol Rev. 2000; 52:557–594. PMID: 11121510.7. Giebisch G, Krapf R, Wagner C. Renal and extrarenal regulation of potassium. Kidney Int. 2007; 72:397–410. PMID: 17568786.
Article9. Hebert SC, Desir G, Giebisch G, Wang W. Molecular diversity and regulation of renal potassium channels. Physiol Rev. 2005; 85:319–371. PMID: 15618483.
Article10. Lin SH, Cheema-Dhadli S, Gowrishankar M, Marliss EB, Kamel KS, Halperin ML. Control of excretion of potassium: lessons from studies during prolonged total fasting in human subjects. Am J Physiol. 1997; 273:F796–F800. PMID: 9374844.11. Kamel KS, Quaggin S, Scheich A, Halperin ML. Disorders of potassium homeostasis: an approach based on pathophysiology. Am J Kidney Dis. 1994; 24:597–613. PMID: 7942818.
Article12. Lin SH, Lin YF, Chen DT, Chu P, Hsu CW, Halperin ML. Laboratory tests to determine the cause of hypokalemia and paralysis. Arch Intern Med. 2004; 164:1561–1566. PMID: 15277290.
Article13. Alazami M, Lin SH, Cheng CJ, Davids MR, Halperin ML. Unusual causes of hypokalaemia and paralysis. QJM. 2006; 99:181–192. PMID: 16469765.
Article14. Lin SH, Lin YF, Halperin ML. Hypokalaemia and paralysis. QJM. 2001; 94:133–139. PMID: 11259688.
Article16. Lin SH, Hsu YD, Cheng NL, Kao MC. Skeletal muscle dihydropyridine-sensitive calcium channel (CACNA1S) gene mutations in chinese patients with hypokalemic periodic paralysis. Am J Med Sci. 2005; 329:66–70. PMID: 15711422.
Article17. Venance SL, Cannon SC, Fialho D, et al. The primary periodic paralyses: diagnosis, pathogenesis and treatment. Brain. 2006; 129:8–17. PMID: 16195244.
Article18. Matthews E, Labrum R, Sweeney MG, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis. Neurology. 2009; 72:1544–1547. PMID: 19118277.
Article19. Andelfinger G, Tapper AR, Welch RC, Vanoye CG, George AL Jr, Benson DW. KCNJ2 mutation results in Andersen syndrome with sex-specific cardiac and skeletal muscle phenotypes. Am J Hum Genet. 2002; 71:663–668. PMID: 12148092.
Article20. Fontaine B, Fournier E, Sternberg D, Vicart S, Tabti N. Hypokalemic periodic paralysis: a model for a clinical and research approach to a rare disorder. Neurotherapeutics. 2007; 4:225–232. PMID: 17395132.
Article21. Holmberg C, Perheentupa J, Launiala K. Colonic electrolyte transport in health and in congenital chloride diarrhea. J Clin Invest. 1975; 56:302–310. PMID: 1150872.
Article22. Hoglund P, Haila S, Socha J, et al. Mutations of the Downregulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat Genet. 1996; 14:316–319. PMID: 8896562.
Article23. Bates CM, Baum M, Quigley R. Cystic fibrosis presenting with hypokalemia and metabolic alkalosis in a previously healthy adolescent. J Am Soc Nephrol. 1997; 8:352–355. PMID: 9048354.
Article24. Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001; 104:545–556. PMID: 11239411.
Article25. Lifton RP, Dluhy RG, Powers M, et al. A chimaeric 11 beta-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. 1992; 355:262–265. PMID: 1731223.26. New MI. Diagnosis and management of congenital adrenal hyperplasia. Annu Rev Med. 1998; 49:311–328. PMID: 9509266.
Article27. Zachmann M. Defects in steroidogenic enzymes. Discrepancies between clinical steroid research and molecular biology results. J Steroid Biochem Mol Biol. 1995; 53:159–164. PMID: 7626448.
Article29. Mune T, Rogerson FM, Nikkilä H, Agarwal AK, White PC. Human hypertension caused by mutations in the kidney isozyme of 11 beta-hydroxysteroid dehydrogenase. Nat Genet. 1995; 10:394–399. PMID: 7670488.30. Lin SH, Yang SS, Chau T, Halperin ML. An unusual cause of hypokalemic paralysis: chronic licorice ingestion. Am J Med Sci. 2003; 325:153–156. PMID: 12640291.
Article31. Kamel KS, Ethier J, Levin A, Halperin ML. Hypokalemia in the "beautiful people". Am J Med. 1990; 88:534–536. PMID: 2186627.
Article32. Chou CL, Chen YH, Chau T, Lin SH. Acquired Bartter-like syndrome associated with gentamicin administration. Am J Med Sci. 2005; 329:144–149. PMID: 15767821.
Article33. Bettinelli A, Bianchetti MG, Girardin E, et al. Use of calcium excretion values to distinguish two forms of primary renal tubular hypokalemic alkalosis: Bartter and Gitelman syndromes. J Pediatr. 1992; 120:38–43. PMID: 1731022.
Article34. Lin SH, Shiang JC, Huang CC, Yang SS, Hsu YJ, Cheng CJ. Phenotype and genotype analysis in Chinese patients with Gitelman's syndrome. J Clin Endocrinol Metab. 2005; 90:2500–2507. PMID: 15687331.
Article35. Peters M, Jeck N, Reinalter S, et al. Clinical presentation of genetically defined patients with hypokalemic salt-losing tubulopathies. Am J Med. 2002; 112:183–190. PMID: 11893344.
Article36. Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005; 85:423–493. PMID: 15788703.
Article37. Bockenhauer D, Feather S, Stanescu HC, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med. 2009; 360:1960–1970. PMID: 19420365.38. Scholl UI, Choi M, Liu T, et al. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci U S A. 2009; 106:5842–5847. PMID: 19289823.
Article39. Pluznick JL, Sansom SC. BK channels in the kidney: role in K(+) secretion and localization of molecular components. Am J Physiol Renal Physiol. 2006; 291:F517–F529. PMID: 16774904.40. Bailey MA, Cantone A, Yan Q, et al. Maxi-K channels contribute to urinary potassium excretion in the ROMK-deficient mouse model of Type II Bartter's syndrome and in adaptation to a high-K diet. Kidney Int. 2006; 70:51–59. PMID: 16710355.
Article41. Kamel KS, Briceno LF, Sanchez MI, et al. A new classification for renal defects in net acid excretion. Am J Kidney Dis. 1997; 29:136–146. PMID: 9002543.
Article42. Igarashi T, Inatomi J, Sekine T, et al. Mutations in SLC4A4 cause permanent isolated proximal renal tubular acidosis with ocular abnormalities. Nat Genet. 1999; 23:264–266. PMID: 10545938.
Article43. Karet FE. Inherited distal renal tubular acidosis. J Am Soc Nephrol. 2002; 13:2178–2184. PMID: 12138152.
Article44. Wrong O, Bruce LJ, Unwin RJ, Toye AM, Tanner MJ. Band 3 mutations, distal renal tubular acidosis, and Southeast Asian ovalocytosis. Kidney Int. 2002; 62:10–19. PMID: 12081559.
Article45. Borthwick KJ, Kandemir N, Topaloglu R, et al. A phenocopy of CAII deficiency: a novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J Med Genet. 2003; 40:115–121. PMID: 12566520.
Article46. Manis JP. Knock out, knock in, knock down--genetically manipulated mice and the Nobel Prize. N Engl J Med. 2007; 357:2426–2429. PMID: 18077807.
