Diagnosis of interstitial lung diseases: from Averill A. Liebow to artificial intelligence
- Affiliations
-
- 1Division of Anatomic Pathology, Mayo Clinic Rochester, Rochester, MN, USA
- 2Division of Anatomic Pathology, Mayo Clinic Arizona, Arizona, FL, USA
- 3Division of Pulmonary and Critical Medicine, Mayo Clinic Rochester, Rochester, MN, USA
- KMID: 2550495
- DOI: http://doi.org/10.4132/jptm.2023.11.17
Abstract
- Histopathologic criteria of usual interstitial pneumonia (UIP)/idiopathic pulmonary fibrosis (IPF) were defined over the years and endorsed by leading organizations decades after Dr. Averill A. Liebow first coined the term UIP in the 1960s as a distinct pathologic pattern of fibrotic interstitial lung disease. Novel technology and recent research on interstitial lung diseases with genetic component shed light on molecular pathogenesis of UIP/IPF. Two antifibrotic agents introduced in the mid-2010s opened a new era of therapeutic approaches to UIP/IPF, albeit contentious issues regarding their efficacy, side effects, and costs. Recently, the concept of progressive pulmonary fibrosis was introduced to acknowledge additional types of progressive fibrosing interstitial lung diseases with the clinical and pathologic phenotypes comparable to those of UIP/IPF. Likewise, some authors have proposed a paradigm shift by considering UIP as a stand-alone diagnostic entity to encompass other fibrosing interstitial lung diseases that manifest a relentless progression as in IPF. These trends signal a pendulum moving toward the tendency of lumping diagnoses, which poses a risk of obscuring potentially important information crucial to both clinical and research purposes. Recent advances in whole slide imaging for digital pathology and artificial intelligence technology could offer an unprecedented opportunity to enhance histopathologic evaluation of interstitial lung diseases. However, current clinical practice trends of moving away from surgical lung biopsies in interstitial lung disease patients may become a limiting factor in this endeavor as it would be difficult to build a large histopathologic database with correlative clinical data required for artificial intelligence models.
Keyword
Figure
-
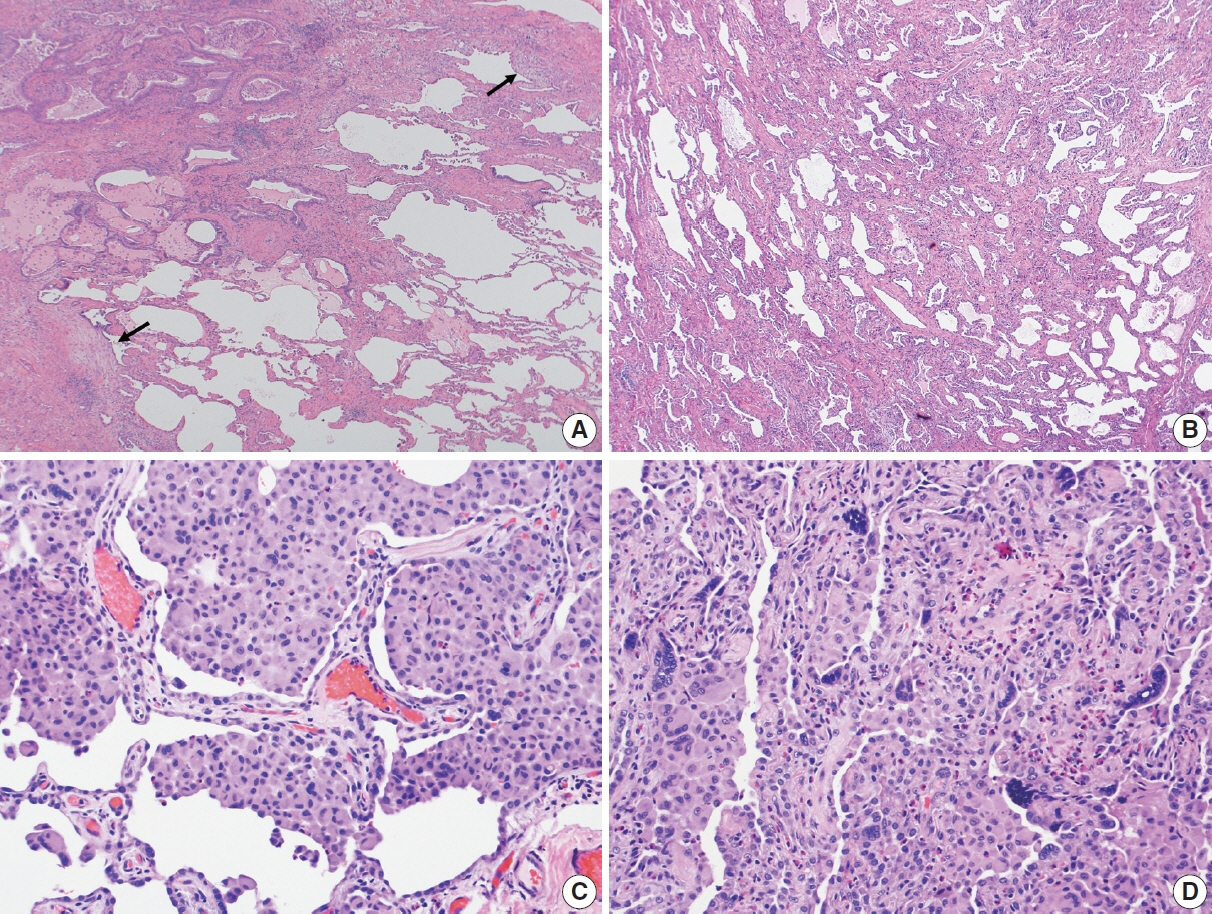
Fig. 1. (A) Usual interstitial pneumonia (UIP), the prototype of interstitial lung disease, showing patchy interstitial fibrosis with subpleural accentuation, marked architectural distortion of alveolar architecture with scarring and microscopic honeycomb changes, and temporal heterogeneity of fibrosis evidenced by scattered fibroblastic foci with dome-shaped myofibroblastic/fibroblastic proliferation over scarred areas (arrows), which likely implies an ongoing acute lung injury in already scarred lung tissue causing progressive clinical course. (B) Nonspecific interstitial pneumonia (NSIP) shows diffuse and uniform fibrous interstitial thickening. In contrast to UIP, NSIP shows relatively well-preserved alveolar architecture without honeycomb changes or fibroblastic foci, which likely explains a more favorable prognosis of NSIP than UIP. (C) Desquamative interstitial pneumonia (DIP) characterized by diffuse collection of pigmented macrophages in the alveolar spaces with mild to moderate interstitial fibrosis. DIP in the original classification by Dr. Liebow is included in the current American Thoracic Society classification. (D) Giant cell interstitial pneumonia (GIP) showing many scattered multinucleated giant cells in the alveolar spaces or septa. GIP was included in the Dr. Liebow’s original classification of interstitial lung diseases but dropped in the current idiopathic interstitial pneumonia classification as GIP is now primarily regarded as a hard metal pneumoconiosis (e.g., cobalt).
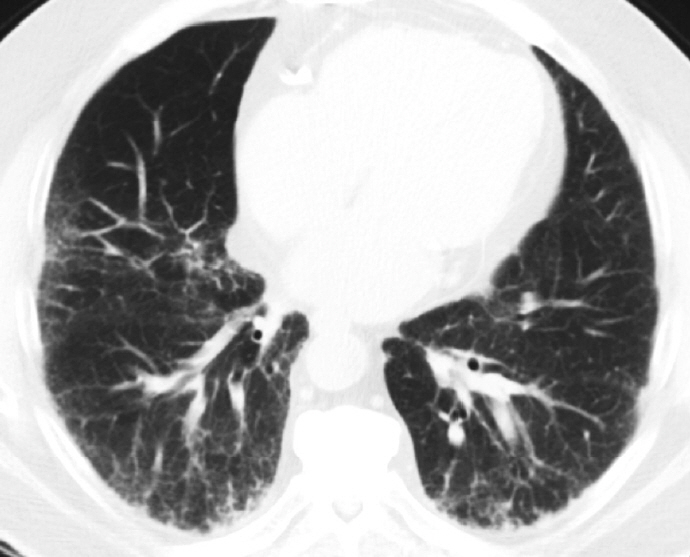
Fig. 2. An axial view of high-resolution computed tomography demonstrates a portion of lower lung fields with classific findings of usual interstitial pneumonia characterized by peripherally accentuated reticular densities and honeycomb changes.
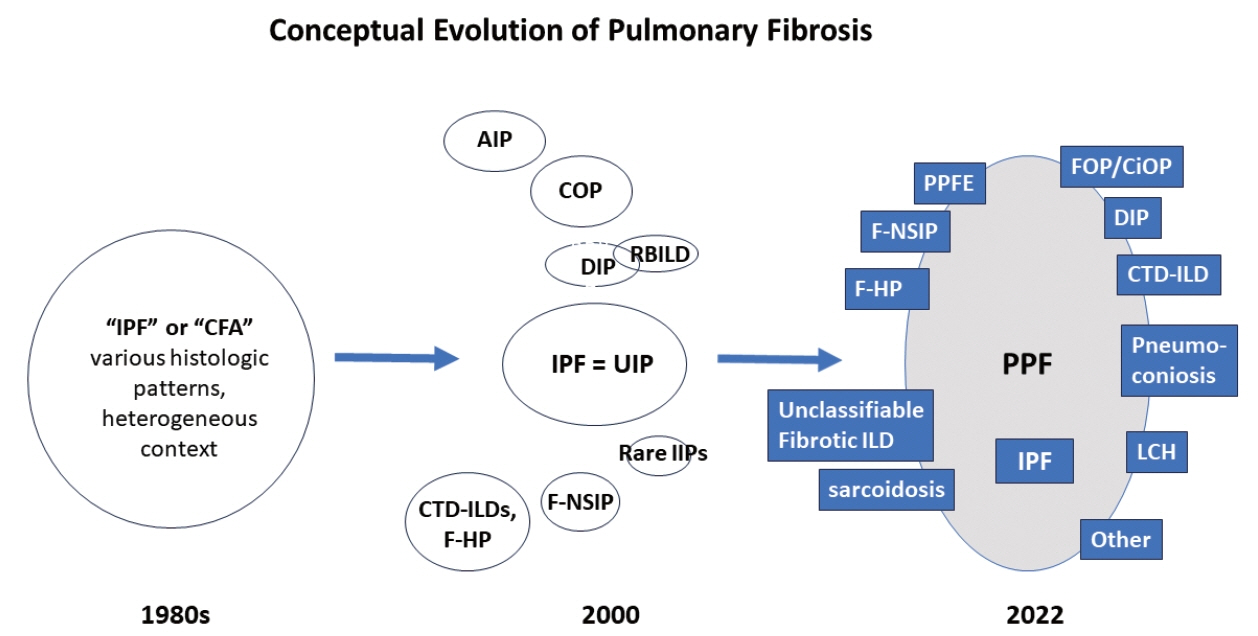
Fig. 3. The idiopathic interstitial pneumonia (IIP) classification established in 2000s is evolved from the less well-defined idiopathic pulmonary fibrosis (IPF) that encompassed various histologic patterns. Progressive pulmonary fibrosis in the current official practice guideline by American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Association (ALAT) can be the manifestion by many fibrotic lung diseases other than IPF. AIP, acute interstitial pneumonia; CFA, cryptogenic fibrosing alveolitis; CiOP, cicatricial organizing pneumonia; COP, cryptogenic organizing pneumonia; CTD-ILD, connective tissue disease–associated interstitial lung disease; DIP, desquamative interstitial pneumonia; F-HP, fibrotic hypersensitivity pneumonitis; F-NSIP, fibrotic nonspecific interstitial pneumonia; FOP, fibrotic organizing pneumonia; LCH, Langerhans cell histiocytosis; PPFE, pleuroparenchymal fibroelastosis; RBILD, respiratory bronchiolitis interstitial lung disease; UIP, usual interstitial pneumonia.
Reference
-
References
1. Liebow A, Carrington CB. The interstitial pneumonias. In : Simon M, Potchen EJ, LeMay M, editors. Frontiers of pulmonary radiology. New York: Grune and Stratton;1969. p. 102–41.2. Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998; 157:1301–15.3. American Thoracic Society. Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am J Respir Crit Care Med. 2000; 161:646–64.4. American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002; 165:277–304.5. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013; 188:733–48.6. Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011; 183:788–824.7. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022; 205:e18–47.8. Kim SY, Diggans J, Pankratz D, et al. Classification of usual interstitial pneumonia in patients with interstitial lung disease: assessment of a machine learning approach using high-dimensional transcriptional data. Lancet Respir Med. 2015; 3:473–82.9. Raghu G, Flaherty KR, Lederer DJ, et al. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med. 2019; 7:487–96.10. Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2007; 176:636–43.11. Azuma A, Nukiwa T, Tsuboi E, et al. Double-blind, placebo-controlled trial of pirfenidone in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2005; 171:1040–7.12. Taniguchi H, Ebina M, Kondoh Y, et al. Pirfenidone in idiopathic pulmonary fibrosis. Eur Respir J. 2010; 35:821–9.13. Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011; 365:1079–87.14. Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014; 370:2071–82.15. Raghu G, Rochwerg B, Zhang Y, et al. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis: an update of the 2011 clinical practice guideline. Am J Respir Crit Care Med. 2015; 192:e3–19.16. Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018; 198:e44–68.17. Selman M, Pardo A, Wells AU. Usual interstitial pneumonia as a stand-alone diagnostic entity: the case for a paradigm shift? Lancet Respir Med. 2023; 11:188–96.18. Hambly N, Farooqi MM, Dvorkin-Gheva A, et al. Prevalence and characteristics of progressive fibrosing interstitial lung disease in a prospective registry. Eur Respir J. 2022; 60:2102571.19. Hozumi H, Nakamura Y, Johkoh T, et al. Acute exacerbation in rheumatoid arthritis-associated interstitial lung disease: a retrospective case control study. BMJ Open. 2013; 3:e003132.20. Kang J, Kim YJ, Choe J, Chae EJ, Song JW. Acute exacerbation of fibrotic hypersensitivity pneumonitis: incidence and outcomes. Respir Res. 2021; 22:152.21. Adegunsoye A, Oldham JM, Bellam SK, et al. Computed tomography honeycombing identifies a progressive fibrotic phenotype with increased mortality across diverse interstitial lung diseases. Ann Am Thorac Soc. 2019; 16:580–8.22. Wijsenbeek M, Cottin V. Spectrum of fibrotic lung diseases. N Engl J Med. 2020; 383:958–68.23. Cottin V, Wollin L, Fischer A, Quaresma M, Stowasser S, Harari S. Fibrosing interstitial lung diseases: knowns and unknowns. Eur Respir Rev. 2019; 28:180100.24. Brown KK, Martinez FJ, Walsh SL, et al. The natural history of progressive fibrosing interstitial lung diseases. Eur Respir J. 2020; 55:2000085.25. Nasser M, Larrieu S, Si-Mohamed S, et al. Progressive fibrosing interstitial lung disease: a clinical cohort (the PROGRESS study). Eur Respir J. 2021; 57:2002718.26. Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ Open Respir Res. 2017; 4:e000212.27. Flaherty KR, Wells AU, Cottin V, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019; 381:1718–27.28. Maher TM, Corte TJ, Fischer A, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. 2020; 8:147–57.29. Behr J, Prasse A, Kreuter M, et al. Pirfenidone in patients with progressive fibrotic interstitial lung diseases other than idiopathic pulmonary fibrosis (RELIEF): a double-blind, randomised, placebocontrolled, phase 2b trial. Lancet Respir Med. 2021; 9:476–86.30. Wells AU, Flaherty KR, Brown KK, et al. Nintedanib in patients with progressive fibrosing interstitial lung diseases-subgroup analyses by interstitial lung disease diagnosis in the INBUILD trial: a randomised, double-blind, placebo-controlled, parallel-group trial. Lancet Respir Med. 2020; 8:453–60.31. Matteson EL, Kelly C, Distler JH, et al. Nintedanib in patients with autoimmune disease-related progressive fibrosing interstitial lung diseases: subgroup analysis of the INBUILD trial. Arthritis Rheumatol. 2022; 74:1039–47.32. Sandoz E. Uber zwei falle von fotaler bronchektasie [About two cases of fetal bronchiectasis]. Beitr Pathol Anat. 1907; 41:496–517.33. Marshall RP, Puddicombe A, Cookson WO, Laurent GJ. Adult familial cryptogenic fibrosing alveolitis in the United Kingdom. Thorax. 2000; 55:143–6.34. Hodgson U, Laitinen T, Tukiainen P. Nationwide prevalence of sporadic and familial idiopathic pulmonary fibrosis: evidence of founder effect among multiplex families in Finland. Thorax. 2002; 57:338–42.35. Barlo NP, van Moorsel CH, Ruven HJ, Zanen P, van den Bosch JM, Grutters JC. Surfactant protein-D predicts survival in patients with idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. 2009; 26:155–61.36. Garcia-Sancho C, Buendia-Roldan I, Fernandez-Plata MR, et al. Familial pulmonary fibrosis is the strongest risk factor for idiopathic pulmonary fibrosis. Respir Med. 2011; 105:1902–7.37. Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol. 2003; 29:S47–50.38. Fernandez BA, Fox G, Bhatia R, et al. A Newfoundland cohort of familial and sporadic idiopathic pulmonary fibrosis patients: clinical and genetic features. Respir Res. 2012; 13:64.39. Ley B, Newton CA, Arnould I, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017; 5:639–47.40. Newton CA, Oldham JM, Ley B, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. 2019; 53:1801641.41. Cutting CC, Bowman WS, Dao N, et al. Family history of pulmonary fibrosis predicts worse survival in patients with interstitial lung disease. Chest. 2021; 159:1913–21.42. Steele MP, Speer MC, Loyd JE, et al. Clinical and pathologic features of familial interstitial pneumonia. Am J Respir Crit Care Med. 2005; 172:1146–52.43. Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016; 48:1710–20.44. Mathai SK, Humphries S, Kropski JA, et al. MUC5B variant is associated with visually and quantitatively detected preclinical pulmonary fibrosis. Thorax. 2019; 74:1131–9.45. Hunninghake GM, Quesada-Arias LD, Carmichael NE, et al. Interstitial lung disease in relatives of patients with pulmonary fibrosis. Am J Respir Crit Care Med. 2020; 201:1240–8.46. Salisbury ML, Hewlett JC, Ding G, et al. Development and progression of radiologic abnormalities in individuals at risk for familial interstitial lung disease. Am J Respir Crit Care Med. 2020; 201:1230–9.47. Mathai SK, Newton CA, Schwartz DA, Garcia CK. Pulmonary fibrosis in the era of stratified medicine. Thorax. 2016; 71:1154–60.48. Seibold MA, Wise AL, Speer MC, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2011; 364:1503–12.49. van Moorsel CH, van der Vis JJ, Grutters JC. Genetic disorders of the surfactant system: focus on adult disease. Eur Respir Rev. 2021; 30:200085.50. Nogee LM, Dunbar AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001; 344:573–9.51. Thomas AQ, Lane K, Phillips J 3rd, et al. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Respir Crit Care Med. 2002; 165:1322–8.52. van Moorsel CH, Ten Klooster L, van Oosterhout MF, et al. SFTPA2 mutations in familial and sporadic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2015; 192:1249–52.53. Armanios MY, Chen JJ, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007; 356:1317–26.54. Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007; 104:7552–7.55. Armanios M. Telomerase and idiopathic pulmonary fibrosis. Mutat Res. 2012; 730:52–8.56. Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin North Am. 2009; 23:215–31.57. Diaz de Leon A, Cronkhite JT, Yilmaz C, et al. Subclinical lung disease, macrocytosis, and premature graying in kindreds with telomerase (TERT) mutations. Chest. 2011; 140:753–63.58. Borie R, Tabeze L, Thabut G, et al. Prevalence and characteristics of TERT and TERC mutations in suspected genetic pulmonary fibrosis. Eur Respir J. 2016; 48:1721–31.59. Juge PA, Borie R, Kannengiesser C, et al. Shared genetic predisposition in rheumatoid arthritis-interstitial lung disease and familial pulmonary fibrosis. Eur Respir J. 2017; 49:1602314.60. Petrovski S, Todd JL, Durheim MT, et al. An exome sequencing study to assess the role of rare genetic variation in pulmonary fibrosis. Am J Respir Crit Care Med. 2017; 196:82–93.61. Dressen A, Abbas AR, Cabanski C, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med. 2018; 6:603–14.62. Ley B, Torgerson DG, Oldham JM, et al. Rare protein-altering telomere-related gene variants in patients with chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2019; 200:1154–63.63. Cronkhite JT, Xing C, Raghu G, et al. Telomere shortening in familial and sporadic pulmonary fibrosis. Am J Respir Crit Care Med. 2008; 178:729–37.64. Leslie KO, Cool CD, Sporn TA, et al. Familial idiopathic interstitial pneumonia: histopathology and survival in 30 patients. Arch Pathol Lab Med. 2012; 136:1366–76.65. Viswanathan VS, Toro P, Corredor G, Mukhopadhyay S, Madabhushi A. The state of the art for artificial intelligence in lung digital pathology. J Pathol. 2022; 257:413–29.66. Makela K, Mayranpaa MI, Sihvo HK, et al. Artificial intelligence identifies inflammation and confirms fibroblast foci as prognostic tissue biomarkers in idiopathic pulmonary fibrosis. Hum Pathol. 2021; 107:58–68.67. Uegami W, Bychkov A, Ozasa M, et al. MIXTURE of human expertise and deep learning-developing an explainable model for predicting pathological diagnosis and survival in patients with interstitial lung disease. Mod Pathol. 2022; 35:1083–91.68. Testa LC, Jule Y, Lundh L, et al. Automated digital quantification of pulmonary fibrosis in human histopathology specimens. Front Med (Lausanne). 2021; 8:607720.69. Ashcroft T, Simpson JM, Timbrell V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J Clin Pathol. 1988; 41:467–70.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Using Artificial Intelligence Software for Diagnosing Emphysema and Interstitial Lung Disease
- Artificial Intelligence in Pathology
- Application of artificial intelligence for diagnosis of early gastric cancer based on magnifying endoscopy with narrow-band imaging
- A Narrative Review on the Application of Artificial Intelligence on the Diagnosis and Outcome Prediction for Spinal Diseases
- The Role of a Diabetologist in the New Era of Artificial Intelligence
