Ann Pediatr Endocrinol Metab.
2023 Dec;28(4):312-317. 10.6065/apem.2244294.147.
Short stature with low serum alkaline phosphatase activity: a case report of hypophosphatasia
- Affiliations
-
- 1Department of Pediatrics, Keimyung University Dongsan Hospital, Keimyung University School of Medicine, Daegu, Korea
- 2Department of Pediatrics, Keimyung University Daegu Dongsan Hospital, Daegu, Korea
- KMID: 2549326
- DOI: http://doi.org/10.6065/apem.2244294.147
Abstract
- Hypophosphatasia (HPP) is a rare condition characterized by abnormal bone mineralization. The manifestations of HPP vary from no symptoms to intrauterine fetal death; short stature is another indication of HPP. A 3 ½-year-old boy presented with short stature, transient hypercalcemia, and mild gait disturbance without definite bony deformity. Laboratory examination revealed transient hypercalcemia, normal phosphorous and 25-hydroxy vitamin D levels, and mildly low alkaline phosphatase levels. A targeted next-generation sequencing panel associated with inborn errors of metabolism revealed a pathogenic heterozygous mutation in the ALPL gene, c.979T>C (p.Phe327Leu). When a child visits a hospital with short stature, decreased height velocity, and low alkaline phosphatase level, clinicians should consider the possibility of HPP even if definite skeletal dysplasia is not evident.
Figure
-
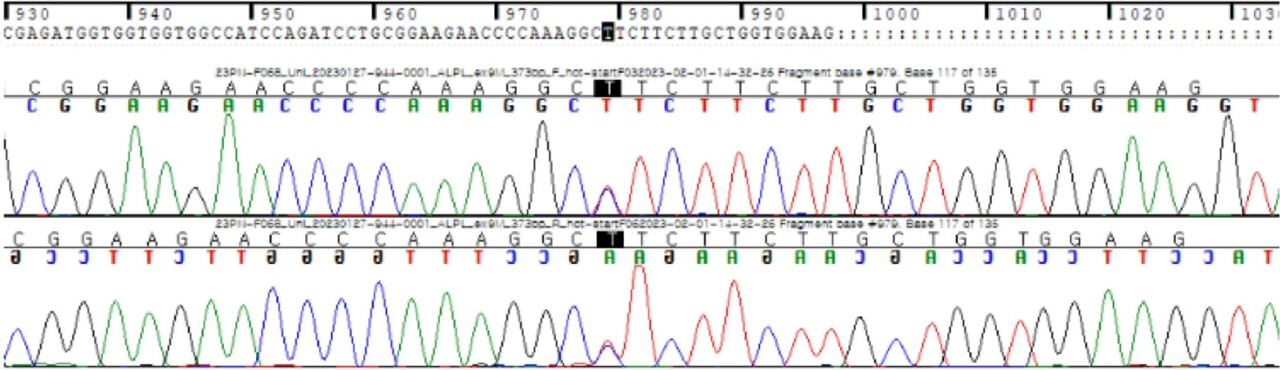
Fig. 1. A heterozygous mutation in the ALPL gene, c.979T>C (p.Phe327Leu).
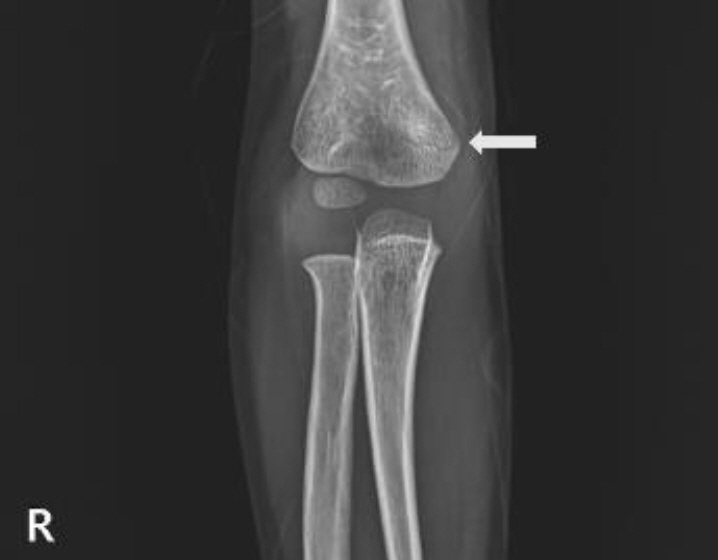
Fig. 2. Humerus radiographs showing a right humerus supracondylar fracture at 3 years 11 months of age (white arrow).
Reference
-
References
1. Mornet E. Hypophosphatasia. Metabolism. 2018; 82:142–55.2. Silvent J, Gasse B, Mornet E, Sire JY. Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J Biol Chem. 2014; 289:24168–79.3. Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001; 281:C1–11.4. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. 2016; 12:233–46.5. Whyte MP, Zhang F, Wenkert D, McAlister WH, Mack KE, Benigno MC, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone. 2015; 75:229–39.6. Taketani T, Onigata K, Kobayashi H, Mushimoto Y, Fukuda S, Yamaguchi S. Clinical and genetic aspects of hypophosphatasia in Japanese patients. Arch Dis Child. 2014; 99:211–5.7. Glotov OS, Savostyanov KV, Nagornova TS, Chernov AN, Fedyakov MA, Raspopova AN, et al. Clinical and genetic characteristics of pediatric patients with hypophosphatasia in the Russian Population. Int J Mol Sci. 2022; 23:12976.8. Colantonio DA, Kyriakopoulou L, Chan MK, Daly CH, Brinc D, Venner AA, et al. Closing the gaps in pediatric laboratory reference intervals: a CALIPER database of 40 biochemical markers in a healthy and multiethnic population of children. Clin Chem. 2012; 58:854–68.9. Whyte MP. Hypophosphatasia: an overview for 2017. Bone. 2017; 102:15–25.10. Buchanan AM, Fiorillo SP, Omondi MW, Cunningham CK, Crump JA. Establishment of biochemistry reference values for healthy Tanzanian infants, children and adolescents in Kilimanjaro region. Trop Med Int Health. 2015; 20:1569–77.11. Estey MP, Cohen AH, Colantonio DA, Chan MK, Marvasti TB, Randell E, et al. CLSI-based transference of the CALIPER database of pediatric reference intervals from Abbott to Beckman, Ortho, Roche and Siemens Clinical Chemistry Assays: direct validation using reference samples from the CALIPER cohort. Clin Biochem. 2013; 46:1197–219.12. Michigami T, Uchihashi T, Suzuki A, Tachikawa K, Nakajima S, Ozono K. Common mutations F310L and T1559del in the tissue-nonspecific alkaline phosphatase gene are related to distinct phenotypes in Japanese patients with hypophosphatasia. Eur J Pediatr. 2005; 164:277–82.13. Demirbilek H, Alanay Y, Alikasifoglu A, Topcu M, Mornet E, Gonc N, et al. Hypophosphatasia presenting with pyridoxine-responsive seizures, hypercalcemia, and pseudotumor cerebri: case report. J Clin Res Pediatr Endocrinol. 2012; 4:34–8.14. Thakker RV, Whyte MP, Eisman JA, Igarashi T. Genetics of bone biology and skeletal disease. 2nd ed. London: Academic Press;2017.15. Offiah AC, Vockley J, Munns CF, Murotsuki J. Differential diagnosis of perinatal hypophosphatasia: radiologic perspectives. Pediatr Radiol. 2019; 49:3–22.16. Michigami T, Tachikawa K, Yamazaki M, Kawai M, Kubota T, Ozono K. Hypophosphatasia in Japan: ALPL mutation analysis in 98 unrelated patients. Calcif Tissue Int. 2020; 106:221–31.17. Okawa R, Kokomoto K, Kitaoka T, Kubota T, Watanabe A, Taketani T, et al. Japanese nationwide survey of hypophosphatasia reveals prominent differences in genetic and dental findings between odonto and non-odonto types. PLoS One. 2019; 14:e0222931.18. Whyte MP, Kurtzberg J, McAlister WH, Mumm S, Podgornik MN, Coburn SP, et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003; 18:624–36.19. Whyte MP, Rockman-Greenberg C, Ozono K, Riese R, Moseley S, Melian A, et al. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J Clin Endocrinol Metab. 2016; 101:334–42.
- Full Text Links
-
- Actions
-
Cited
- CITED
-
- Close
- Share
-
- Similar articles
-
- Case Report of Lethal Perinatal Hypophosphatasia with Seizure and Respiratory Failure Diagnosed by ALPL Gene Mutation
- Serum alkaline phosphatase activity after intravenous administration of albumin preparation
- Bilateral Transverse (Bowdler) Fibular Spurs with Hypophosphatasia in an Adolescent Girl
- Musculoskeletal and neurocognitive clinical significance of adult hypophosphatasia
- Studies on Alkaline Phosphatase Isoenzyme in the Serum and Organs of the Rat
